2 个不稳定版本
| 0.2.0 | 2023年12月29日 |
|---|---|
| 0.1.0 | 2023年11月20日 |
#139 in 生物学
160KB
408 行
to-trans
从 fasta + GTF/GFF 构建高性能外显子/编码区剪接转录组。这是一个用 Rust 编写的命令行工具,通过使用基因组 (.fa) 和基因模型 (.gtf/.gff) 来构建转录组。
用法
High-performance transcriptome builder from fasta + GTF/GFF
Usage: to-trans --fasta <FASTA> --gtf <GTF> [OPTIONS]
--Arguments:
-f, --fasta <FASTA> Path to your .fa file
-g, --gtf <GTF/GFF> Path to your .gtf/.gff file
Options:
-m, --mode <MODE> Feature to extract from GTF/GFF file (exon or CDS) [default: exon]
-o, --out <OUT> Path to output file [default: transcriptome.fa].
-t, --threads <THREADS> Number of threads [default: max ncpus]
-h, --help Print help
-V, --version Print version
crate: https://crates.io/crates/to-trans
版本 0.2.0 的新特性
- 现在 to-trans 的速度提高了约 2-3 秒!
- 并行方法现在是组装转录序列的主算法
即将推出...
to-trans 将随着时间的推移不断成长,扩展其选项和功能。在下一个版本中,将推出以下功能:内含子提取、基于长度的转录组、针对特定染色体的构建,以及其他功能!
安装/构建
要安装 to-trans,请执行以下操作
- 获取 Rust:在 Unix 上,请运行
curl https://sh.rustup.rs -sSf | sh,或者访问 此处 获取其他选项 - 运行
cargo install to-trans(确保在运行之前将~/.cargo/bin添加到您的$PATH环境变量中)
要构建 to-trans,请执行以下操作
- 获取 Rust(如上所述)
- 运行
git clone https://github.com/alejandrogzi/to-trans.git && cd to-trans - 运行
cargo run --release <FASTA> <GTF/GFF> <MODEL> <OUTPUT>
默认情况下,to-trans使用exon模式,并将输出发送到./transcriptome.fa
基准测试
请注意,这个基准测试已经过时了。现在to-trans快2-3秒!对于人类基因组/gtf,to-trans构建完整的转录组需要6秒,这大约是GFFRead的3倍快!
除了某些特定物种,例如人类(GRCh38)或小鼠(GRCm39)拥有可用的转录组外,大多数动物界都没有预先定义的转录序列文件。当在转录/异构体层面上工作时,这成为一个问题。
与GFFRead(1)相比,这是一个功能强大的gff/gtf工具,to-trans可以在不需要索引输入基因组的情况下,2倍快地构建完整的转录组。在人类模型上,to-trans达到最多8秒,而GFFRead最多15秒(已经有一个索引.fai)。对于狗,一个在公共数据库中未提供转录序列的物种,to-trans需要3.5秒,而GFFRead加倍(分别为6秒和12秒,分别对应于已索引和未索引的基因组)。
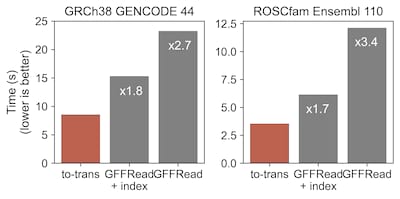
to-trans提供了一种新颖的选项,可以从基因组+基因模型高效地构建转录组。这个工具提供了高性能和效率,无需环境或复杂的依赖关系,并且可以轻松地附加到工作流程/管道中。
参考文献
- Pertea G和Pertea M. GFF Utilities: GffRead和GffCompare [版本1;同行评审:3篇批准]。F1000Research 2020,9:304 (https://doi.org/10.12688/f1000research.23297.1) https://github.com/gpertea/gffread
依赖关系
~3–15MB
~140K SLoC