12 个版本
| 0.4.0-alpha.0 | 2020 年 10 月 19 日 |
|---|---|
| 0.3.2 | 2023 年 8 月 7 日 |
| 0.3.1 | 2020 年 10 月 18 日 |
| 0.3.0 | 2018 年 8 月 21 日 |
| 0.1.0 | 2017 年 7 月 31 日 |
在 解析实现 中排名 136
每月下载 687 次
在 26 个 crate 中使用 (23 个直接使用)
255KB
5K SLoC
Rust 中的 FASTA 和 FASTQ 解析和写入。
![]()

注意: master 分支包含 0.4.0 版本的开发。当前稳定版本在 单独的分支 上。
此库提供了以下序列格式的读取器
- FASTA
- FASTQ (包括多行 FASTQ)
- "FASTX":自动识别序列格式(FASTA 或 FASTQ)
特性
- 快速读取器,最小化内存分配和复制使用
- 灵活的写入 FASTA 和 FASTQ 的方法
- 包含精确位置信息的错误信息
- 支持记录位置和回溯
- Serde 支持(对于拥有数据结构)
- 并行处理函数
- 使用模糊测试技术彻底测试 见此处
简单示例
从 STDIN 读取 FASTA 序列,如果其长度大于 100,则将其写入 STDOUT。否则,它将打印一条消息。
use seq_io::fasta::{Reader,Record};
use std::io;
let mut reader = Reader::new(io::stdin());
let mut stdout = io::stdout();
while let Some(result) = reader.next() {
let record = result.expect("reading error");
// determine sequence length
let seqlen = record.seq_lines()
.fold(0, |l, seq| l + seq.len());
if seqlen > 100 {
record.write_wrap(&mut stdout, 80).expect("writing error");
} else {
eprintln!("{} is only {} long", record.id().expect("not UTF-8"), seqlen);
}
}
记录直接从内部缓冲读取器借用数据,不进行进一步的分配或复制。因此,必须使用 while 构造而不是 for 循环。
seq_lines 直接遍历序列行,记录记住其位置,再次不进行复制。
注意: 请确保在 Cargo.toml 中的发布配置文件中添加 lto = true,因为否则不会内联对底层缓冲读取器函数的调用(buf_redux)。
Rust 中的类似项目
- Rust-Bio:提供简单且易于使用的 FASTA 和 FASTQ 读取器的生物信息学库。
- fastq-rs:非常快的 FASTQ 解析器。在许多方面,
seq_io都受到了fastq_rs的启发。 - Needletail:FASTA、FASTQ、FASTX
- fasten 实现了自己的 FASTQ 读取器
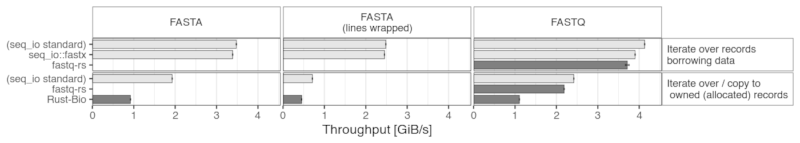
性能比较
以下柱状图显示了在内存中生成的随机序列(FASTA 序列可能在一行中,或者缩进到 80 个字符宽度)的一些基准测试结果。
此包中的读取器还与 fastq-rs 和 Rust-Bio 解析器进行了比较。后者仅在“所有者”部分存在,因为没有迭代记录而不分配记录的可能性。
更多基准测试可以在 单独的页面 上找到。
依赖关系
~1–2MB
~37K SLoC