4 个版本
| 0.2.2 | 2024 年 3 月 1 日 |
|---|---|
| 0.2.1 | 2023 年 12 月 16 日 |
| 0.1.1 | 2023 年 10 月 19 日 |
| 0.1.0 | 2023 年 9 月 12 日 |
#211 in 科学
86 每月下载量
595KB
654 行
gtfsort
使用基于字典序索引排序算法优化的 chr/pos/feature GTF/GFF 排序器。
- 现在支持 GFF 文件!
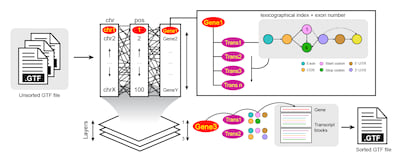
虽然当前的工具(大部分集中在 GFF3)已被推荐用于排序 GTF 文件,但没有一个专门针对 chr/pos/feature 排序。这种方法确保了自定义排序方向,这对于减少使用排序 GTF 文件的工具的计算时间非常有用。此外,它提供了友好且有序的基因结构可视化(基因 -> 转录 -> CDS/exon -> 起始/终止 -> UTR/Sel),使用户能够更有效地搜索特征。
[!注意]
如果您在您的工作中使用了 gtfsort,请引用
Gonzales-Irribarren A. gtfsort:一种高效排序 GTF 文件的工具。bioRxiv 2023.10.21.563454; doi: https://doi.org/10.1101/2023.10.21.563454
用法
Usage: gtfsort -i <GTF> -o <OUTPUT> [-t <THREADS>]
Arguments:
-i, --input <GTF>: unsorted GTF file
-o, --output <OUTPUT>: sorted GTF file
Options:
-t, --threads <THREADS>: number of threads [default: your max ncpus]
--help: print help
--version: print version
版本 0.2.2 的新功能
- gtfsort 现在支持 GFF 排序!
- gtfsort 现在更快(约 0.2 秒);1.9GB (Cyprinus carpio carpio) 在 6.7 秒内
- 部分代码已被重新组织并改进。
- nf 模块和 galaxy 工具即将推出!(这些正在制作中)
crate: https://crates.io/crates/gtfsort
点击查看详细格式
GTF 代表基因转移格式。GTF 格式是一种 9 列文本格式,用于描述和表示基因组特征。GTF 文件中的每一列都代表有用的信息 [1]
<seqname>
The <seqname> field contains the name of the sequence which this gene is on.
<source>
The <source> field should be a unique label indicating where the annotations came from – typically the name of either a prediction program or a public database.
<feature>
The <feature> field can take 4 values: "CDS", "start_codon", "stop_codon" and "exon". The “CDS” feature represents the coding sequence starting with the first translated codon and proceeding to the last translated codon. Unlike Genbank annotation, the stop codon is not included in the “CDS” feature for the terminal exon. The “exon” feature is used to annotate all exons, including non-coding exons. The “start_codon” and “stop_codon” features should have a total length of three for any transcript but may be split onto more than one line in the rare case where an intron falls inside the codon.
<start>, <end>
Integer start and end coordinates of the feature relative to the beginning of the sequence named in <seqname>. <start> must be less than or equal to <end>. Sequence numbering starts at 1. Values of <start> and <end> must fall inside the sequence on which this feature resides.
<score>
The <score> field is used to store some score for the feature. This can be any numerical value, or can be left out and replaced with a period.
<strand>
'+' or '-'.
<frame>
A value of 0 indicates that the first whole codon of the reading frame is located at 5'-most base. 1 means that there is one extra base before the first whole codon and 2 means that there are two extra bases before the first whole codon. Note that the frame is not the length of the CDS mod 3. If the strand is '-', then the first base of the region is value of <end>, because the corresponding coding region will run from <end> to <start> on the reverse strand.
<attributes>
Each attribute in the <attribute> field should have the form: attribute_name “attribute_value”;
Attributes must end in a semicolon which must then be separated from the start of any subsequent attribute by exactly one space character (NOT a tab character). Attributes’ values should be surrounded by double quotes.
GTF 格式有不同的版本,最常用的是 GTF2.5 和 GTF3(基于 Ensembl 的结构)。每个版本与其他版本的主要区别在于属性中的特征排序。gtfsort 设计用于与 GTF2.5 和 GTF3 一起工作。
| 格式 | ... | 特征 | ... | 属性 |
|---|---|---|---|---|
| GTF2.5 | ... | 基因,转录本,外显子,CDS,UTR,起始密码子,终止密码子,硒代半胱氨酸 | ... | 属性名“属性值”;属性名“属性值”; |
| GTF3 | ... | 基因,转录本,外显子,CDS,硒代半胱氨酸,起始密码子,终止密码子,3'UTR 和 5'UTR | ... | 属性名“属性值”;属性名“属性值”; |
安装
在您的系统上安装 gtfsort 的步骤如下
- 获取 Rust:在 Unix 上使用
curl https://sh.rustup.rs -sSf | sh,或访问 此处 获取其他选项 - 运行
cargo install gtfsort(确保在运行之前~/.cargo/bin已添加到您的$PATH中) - 使用
gtfsort并提供所需的参数
构建
要从该仓库构建 gtfsort,请执行以下操作
- 获取 rust(如上所述)
- 运行
git clone https://github.com/alejandrogzi/gtfsort.git && cd gtfsort - 运行
cargo run --release -- -i <GTF> -o <OUTPUT>
容器镜像
要构建开发容器镜像
- 运行
git clone https://github.com/alejandrogzi/gtfsort.git && cd gtfsort - 使用
start docker或systemctl start docker初始化 docker - 构建镜像
docker image build --tag gtfsort . - 运行
docker run --rm -v "[dir_where_your_gtf_is]:/dir" gtfsort -/dir/<INPUT> -o /dir/<OUTPUT>
Conda
要使用 Conda 安装 gtfsort,只需
conda install gtfsort -c bioconda或conda create -n gtfsort -c bioconda gtfsort
基准测试
请注意,这个基准测试已经过时,当前实现(v.0.2.1)比之前的一个(v.0.1.1)快2倍。现在,gtfsort可以在 6.2秒 内排序完整的 Homo sapiens GENCODE 44 GTF(1.5GB),并且可以比之前实现快(约14-15秒)地在 6.7秒 内排序完整的 Cyprinus carpio carpio GTF(1.9GB)。
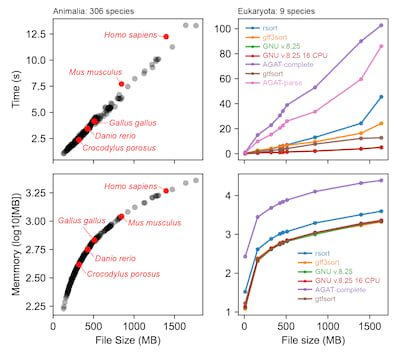
为了评估 gtfsort 的效率和结果,进行了两个主要的基准测试。首先,我在 Ensembl Animalia GTF3 整个数据集(第110次发布;306种物种)上运行了 gtfsort [2]。在这里,gtfsort 展示了它们的主要属性:速度和效率。这个工具能够在使用不到2.5GB RAM的情况下,以高精度在12秒内对1.9GB的GTF文件(Cyprinus carpio carpio)进行排序,并突出显示常见的物种。
其次,我对 gtfsort 工具与几个现有软件工具进行了比较分析:GNU v.8.25(单核和多核配置)、AGAT(在完整和部分解析阶段都使用 --gff 标志)[3]、gff3sort(包括 --precise 和 --chr_order natural 等特定选项)[4]、以及 rsort(一个未发表的具有嵌套数据结构的 Rust 多核实现)。这项综合评估涵盖了广泛的生物领域,包括细菌、真菌、昆虫、哺乳动物等。为了确保评估的稳健性,我使用了九种常见物种:Homo sapiens、Mus musculus、Canis lupus familiaris、Gallus gallus、Danio rerio、Salmo salar、Crocodylus porosus、Drosophila melanogaster 和 Saccharomyces cerevisiae。
在本比较分析中,gtfsort展示了惊人的效率,其计算时间仅次于GNU软件(单核和多核模式),排名第二。然而,值得注意的是,GNU软件无法始终如一地保持染色体/位置/特征的稳定顺序,在排序注释行(例如,以“#”开头的文件开头行)时遇到困难。其余工具的处理时间明显更长,其中一些采用了并行处理方法(例如,rsort,使用了16个核心)。
此外,值得注意的是,在评估的三种工具中,排序文件所需的内存分配在三种工具中保持保守:GNU(单核和多核)、gff3sort和gtfsort。处理最大文件时的内存利用率没有超过2.3 Gbs,即使处理的大型数据集(大小高达1.6 Gbs)。
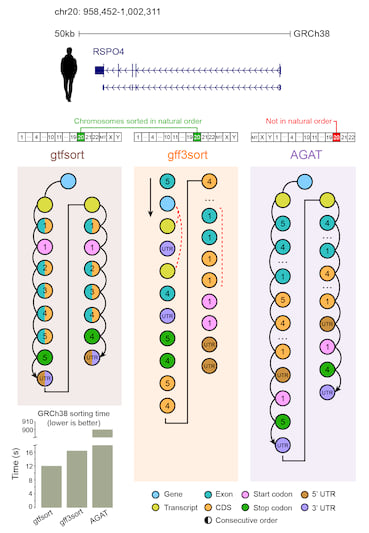
在前一步骤中使用的一系列工具中,只有三种声明包含特征排序步骤[5]:gff3sort、AGAT和gtfsort。gff3sort是一个基于Perl的程序,专门用于排序GFF3/GTF文件,擅长生成与tabix工具[4]兼容的结果。它采用拓扑算法,在初始的两个排序阶段(首先是染色体,然后是位置)之后对特征进行排序。AGAT是一个同样用Perl编写的分析工具包,其中包含一个GFF3/GTF排序工具,在agat_convert_sp_gxf2gxf.pl脚本[3]中,同样采用拓扑排序方法。
为了评估这三个工具的性能,我们将它们应用于最新Ensembl版本(110版本)的GRCh38 Homo sapiens GTF文件。在测试的软件中,gtfsort的速度最快,处理时间为12.0220秒,其次是gff3sort,处理时间为16.3970秒,AGAT需要大约900秒来完成排序操作。AGAT计算时间漫长的著名差异是由于agat_convert_sp_gxf2gxf.pl不仅排序GTF文件,还检查一些有争议的行并修复/添加修正/缺失的行。
尽管计算时间是一个重要的特性,但实际的排序输出将是关键变量。我选择一个随机基因(包括所有其转录物/特征)并测试输出排序是否展示了一个连贯和准确的布局。
- 染色体排序:只有gtfsort和gff3sort展示了直观的排序(从染色体1开始到染色体X结束)。AGAT在这里失败,将MT和性染色体置于开头。
- 特征排序:gff3sort (--precise --chr_order natural) 完全无法展示特征的有序结构(这在块的开始处第一个转录本的exon 5中可以迅速感知)。AGAT和gtfsort相反,确实展示了直观的结构顺序:基因 -> 转录物 -> 特征。AGAT为每个转录物提供2个块,所有CDS都在所有外显子之后,并带有起始/终止密码子和UTR位于末尾。另一方面,gtfsort采用了一种独特的方法,将特征及其相应的外显子编号成对或成组展示,并按降序排序,甚至对负链上的序列也是如此。UTR始终位于序列的末尾,使信息与给定外显子(外显子/CDS/起始/终止)关联的自然和快速理解成为可能。
-
此处所示的所有值代表每个物种连续五次迭代的平均值,包括时间和内存使用。
-
鉴于AGAT-complete和AGAT-parse的显著延长的计算时间,我们已将这些工具的时间值以小数形式(除以10)表示,以便在可视化时提高清晰度。
-
所有基准评估都是在AMD Ryzen 7 5700X和128 GB RAM上进行的。
局限性
gtfsort在公开发布时,仅接受GTF2.5和GTF3格式。允许用户在参数中指定他们的自定义顺序(例如,--parent gene --middle mRNA --child exon,TSS,intron)将很有趣。
参考文献
依赖项
~4–16MB
~152K SLoC