1 个不稳定版本
| 0.4.0 | 2023 年 10 月 6 日 |
|---|
#179 in 生物学
205KB
568 行
![]()
Kractor
kraken extractor
Kractor 根据通过 Kraken2 获得的分类学信息提取序列读数。它消耗 Kraken2 标准输出以及配对或非配对 fastq[.gz/.bz] 文件作为输入。它可以选择消费 Kraken2 报告以提取给定 taxid 的所有分类学父类和子类。默认快速,它输出 fast[q/a] 文件,这些文件可以选择进行压缩。
Kractor 与 KrakenTools 相比,无论是配对还是非配对读数,都显著提高了处理速度。对于压缩的 fastq,配对读数处理速度提高了约 21 倍,对于未压缩的,提高了约 10 倍。非配对读数对于压缩和未压缩输入都提高了约 4 倍。
有关更多信息,请参阅基准测试
动机
深受伟大的 KrakenTools 启发。
在撰写 KrakenTools 时,它作为一个单线程 Python 实现,当处理大型配对端 fastq 文件时,在速度上存在局限性。主要动机是在解析和提取(写入)大量读数时提高速度 - 同时学习 rust!
安装
二进制文件
Linux、MacOS 和 Windows 的预编译二进制文件附在最新版本 0.4.0
Cargo
需要 cargo
cargo install kractor
从源代码构建
安装 rust 工具链
有关安装说明,请参阅 rust 文档: 文档
克隆仓库
git clone https://github.com/Sam-Sims/Kractor
构建并添加到路径
cd Kractor
cargo build --release
export PATH=$PATH:$(pwd)/target/release
所有可执行文件都将位于 Kractor/target/release 目录中。
使用方法
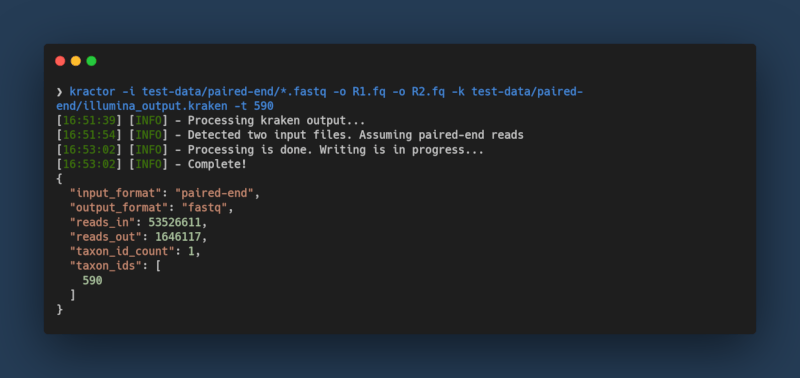
基本使用方法
kractor -k <kraken_output> -i <fastq_file> -t <taxonomic_id> -o <output_file> > kractor_report.json
或者,如果您有配对末端Illumina reads
kractor -k <kraken_output> -i <R1_fastq_file> -i <R2_fastq_file> -t <taxonomic_id> -o <R1_output_file> -o <R2_output_file>
如果您想提取一个分类单元的所有子节点
kractor -k <kraken_output> -r <kraken_report> -i <fastq_file> -t <taxonomic_id> --children -o <output_file>
参数
必需
输入
-i, --输入
此选项将指定包含您想要从中提取读取的输入文件的路径。这些文件可以是压缩的 - (gz, bz2)。可以通过以下方式指定配对末端读取:
使用 --input 两次: -i <R1_fastq_file> -<R2_fastq_file>
使用 --input 一次但传递两个文件: -<R1_fastq_file> <R2_fastq_file>
这意味着bash通配符展开可以工作: -*.fastq
输出
-o, --输出
此选项将指定包含提取读取的输出文件的路径。输出文件的顺序假定与输入相同。
默认情况下,对于支持的文件类型(gz, bz),将推断压缩类型。如果无法推断输出类型,则输出纯文本。
Kraken 输出
-k, --kraken
此选项将指定包含读取ID分类学分类的Kraken2输出路径。
税id
-t, --taxid
此选项将指定您想要从中提取的读取的税id。
可选
输出类型
-O, --输出-类型
此选项将手动设置用于输出文件的压缩模式,并将覆盖从输出路径推断出的类型。
有效值是
gz以输出 gzbz2以输出 bz2none不应用压缩
压缩级别
-l, --级别
此选项将设置压缩输出时使用的压缩级别。应该是1到9之间的值,1为最快但文件大小最大,9为最慢,但文件大小最佳。默认设置为2,因为它在速度/文件大小方面是一个很好的折衷。
输出fasta
--output-fasta
此选项将输出一个fasta文件,读取ID作为标题。
Kraken 报告
-r, --报告
此选项指定由Kraken2生成的报告文件的路径。如果您想使用 --parents 或 --children,则该参数是必需的。
父项
--parents
这将提取在根和指定的 --taxid 之间的所有分类单元的读取。
子项
--children
这将提取所有被分类为 --taxid 的后代或子分类单元的读取(包括taxid)。
排除
--exclude
这将输出除了匹配taxid之外的每个读取。与 --parents 和 --children 一起使用。
跳过报告
--no-json
这将跳过在程序完成时输出到stdout的json报告。
未来计划
- 支持未解压的fastq文件
- 支持配对末端FASTQ文件
-
--include-parents和--include-children参数 - 提供多个分类ID以提取
- 排除分类ID
-
--compression-模式 - 更详细的输出
- 基准
- 输出fasta
- 输出非
gz文件 - 测试
版本
- 0.4.0
变更日志
0.4.0
- Json 报告在成功完成后包含在 stdout 中(可以使用 --no-json 禁用)
- 重命名
0.3.0
- 支持配对末端文件
- 底层主要更改,使用 Noodles 进行 fastq 解析,并使用 Niffler 处理压缩(再见我的代码)
- 使用
--output-fasta输出 fasta 文件 - 简化与压缩类型/级别相关的参数
- 改进日志记录
0.2.3
- 代码优化
0.2.2
- 增加对用户输出的详细程度
--no-compress标志以输出标准纯文本 fastq 文件--exclude排除指定的读取。与--children和--parents一起使用- 底层文档字符串
0.2.1
- 修复以减少内存使用
0.2.0
- 检测和处理
gz文件或普通文件 --compression参数选择压缩类型zlib-ng加速 gzip 处理--children和--parents根据 kraken 报告保存子项和父项
0.1.0
- 首次发布
依赖项
~8–11MB
~180K SLoC