15 个版本
| 0.1.33 | 2023年7月2日 |
|---|---|
| 0.1.31 | 2022年11月8日 |
| 0.1.29 | 2022年3月9日 |
| 0.1.24 | 2021年12月22日 |
| 0.1.3 |
|
#470 在 命令行工具
7MB
2.5K SLoC
rustybam
![]()
![]()
![]()
![]()
rustybam 是一个用 Rust 编程语言编写的生物信息学工具包,专注于操作比对 (bam 和 PAF)、注释 (bed) 和序列 (fasta 和 fastq) 文件。
rustybam 能做什么?
这里有一个注释示例,突出了 rustybam 的某些优点,并演示了如何将每个结果直接读取到另一个子命令。
rb trim-paf .test/asm_small.paf `#trims back alignments that align the same query sequence more than once` \
| rb break-paf --max-size 100 `#breaks the alignment into smaller pieces on indels of 100 bases or more` \
| rb orient `#orients each contig so that the majority of bases are forward aligned` \
| rb liftover --bed <(printf "chr22\t12000000\t13000000\n") `#subsets and trims the alignment to 1 Mbp of chr22.` \
| rb filter --paired-len 10000 `#filters for query sequences that have at least 10,000 bases aligned to a target across all alignments.` \
| rb stats --paf `#calculates statistics from the trimmed paf file` \
| less -S
用法
rustybam [OPTIONS] <SUBCOMMAND>
或
rb [OPTIONS] <SUBCOMMAND>
子命令
子命令的完整手册可以在 文档 中找到。
SUBCOMMANDS:
stats Get percent identity stats from a sam/bam/cram or PAF
bed-length Count the number of bases in a bed file [aliases: bedlen, bl, bedlength]
filter Filter PAF records in various ways
invert Invert the target and query sequences in a PAF along with the CIGAR string
liftover Liftover target sequence coordinates onto query sequence using a PAF
trim-paf Trim paf records that overlap in query sequence [aliases: trim, tp]
orient Orient paf records so that most of the bases are in the forward direction
break-paf Break PAF records with large indels into multiple records (useful for
SafFire) [aliases: breakpaf, bp]
paf-to-sam Convert a PAF file into a SAM file. Warning, all alignments will be marked as
primary! [aliases: paftosam, p2s, paf2sam]
fasta-split Reads in a fasta from stdin and divides into files (can compress by adding
.gz) [aliases: fastasplit, fasplit]
fastq-split Reads in a fastq from stdin and divides into files (can compress by adding
.gz) [aliases: fastqsplit, fqsplit]
get-fasta Mimic bedtools getfasta but allow for bgzip in both bed and fasta inputs
[aliases: getfasta, gf]
nucfreq Get the frequencies of each bp at each position
repeat Report the longest exact repeat length at every position in a fasta
suns Extract the intervals in a genome (fasta) that are made up of SUNs
help Print this message or the help of the given subcommand(s)
安装
conda
mamba install -c bioconda rustybam
cargo
cargo install rustybam
预编译的二进制文件
从 发布 下载(可能比本地编译版本慢)。
源代码
git clone https://github.com/mrvollger/rustybam.git
cd rustybam
cargo build --release
并且可执行文件将在这里构建
target/release/{rustybam,rb}
示例
PAF 或 BAM 统计
对于具有扩展 CIGAR 操作的 BAM 文件,我们可以计算比对的统计信息并以 BED 格式报告。
rustybam stats {input.bam} > {stats.bed}
只要使用 -c --eqx 生成,也可以对 PAF 文件执行相同的操作。
rustybam stats --paf {input.paf} > {stats.bed}
PAF liftovers
我有一个
PAF,我想只为参考中的一个特定区域对其进行子集化。
使用 rustybam 很简单
rustybam liftover \
--bed <(printf "chr1\t0\t250000000\n") \
input.paf > trimmed.paf
但我也想得到该区域的比对统计信息。
没问题,rustybam liftover 不仅裁剪坐标,还裁剪 CIGAR,因此它可以直接用于 rustybam stats
rustybam liftover \
--bed <(printf "chr1\t0\t250000000\n") \
input.paf \
| rustybam stats --paf \
> trimmed.stats.bed
但是,Evan 要求一个“对齐滑块”,所以我需要分块重新对齐。
不需要,只需将你的 bed 查询设置为 rustybam liftoff 的一系列滑动窗口,它就会完成剩下的工作。
rustybam liftover \
--bed <(bedtools makewindows -w 100000 \
<(printf "chr1\t0\t250000000\n") \
) \
input.paf \
| rustybam stats --paf \
> trimmed.stats.bed
你还可以使用 rustybam breakpaf 将大于一定大小的插入/删除的 PAF 记录拆分成更多“miropeats”间隔。
rustybam breakpaf --max-size 1000 input.paf \
| rustybam liftover \
--bed <(printf "chr1\t0\t250000000\n") \
| ./rustybam stats --paf \
> trimmed.stats.bed
但是,我如何可视化数据呢?
试试 SafFire!
一次对齐
在CNVs和倒位边界处,minimap2可能会将查询序列的同一部分与目标序列的多个片段对齐。此实用程序使用PAF对齐的CIGAR字符串(必须使用--eqx)来确定对齐的最佳分割,以确保没有任何查询碱基被对齐多次。为此,整个PAF文件被加载到内存中,然后从最大的重叠区间开始迭代移除重叠。
rb trim-paf {input.paf} > {trimmed.paf}
以下是从NOTCH2NL区域的一个示例,比较修剪前后的CHM1与CHM13: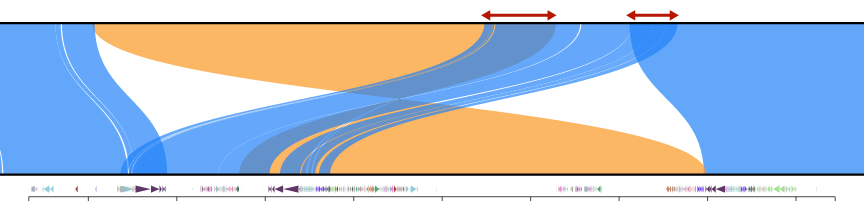
修剪后: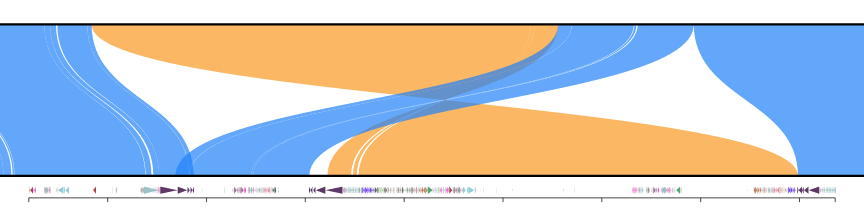
分割fastx文件
在stdout和其他两个(压缩和解压缩)文件之间分割fasta文件。
cat {input.fasta} | rustybam fasta-split two.fa.gz three.fa
在stdout和其他两个(压缩和解压缩)文件之间分割fastq文件。
cat {input.fastq} | rustybam fastq-split two.fq.gz three.fq
从fasta中提取
此工具旨在模仿bedtools getfasta,但此工具允许fasta文件为bgzipped。
samtools faidx {seq.fa(.gz)}
rb get-fasta --name --strand --bed {regions.of.interest.bed} --fasta {seq.fa(.gz)}
待办事项
- 添加一个像
bedtools getfasta一样的操作,它实际上可以与bgzipped输入一起工作。- 实现bed12/split
- 允许使用sam或paf进行操作
- 从PAF文件制作sam标题
- 将sam记录转换为paf记录
- 将paf记录转换为sam记录
- 使工具无缝地与sam和paf一起工作
- 为Nucfreq添加
D4 - 完成实现
suns。 - 在
bed-length中允许多个输入文件 - 开始维护变更日志
依赖关系
~29–40MB
~633K SLoC