1 个不稳定版本
| 新 0.6.9 | 2024年8月17日 |
|---|
#7 in 生物学
127 每月下载次数
595KB
6.5K SLoC
PlasCAD
![]()
![]()
质粒和引物创建及验证的设计软件。
安装
Windows和Linux
Mac
通过下载和安装Rust,然后从命令行运行cargo install plascad来从源代码编译。
当前功能
引物质量控制和调整
在多个指标上评估引物的质量:熔融温度、GC含量、3'端稳定性、重复序列以及形成自身末端二聚体的可能性。
这允许自动或手动更改引物长度以优化这些参数。这是通过标记一个或多个引物端不具固定起始点,并在此端提供超过预期的匹配序列核苷酸来实现的。然后可以调整终点以优化引物质量。
当两端都标记为可调整时,PlasCAD假定此引物是用于连接两个DNA片段,如SLIC和FC克隆。
SLIC和FastCloning的引物生成
给定插入、载体和插入点的序列,它将生成适合SLIC和FastCloning的引物。它生成扩增整个载体的引物,以及包含与载体合适重叠区域的插入引物。
它还会创建一个包含克隆产物序列的新文件。
序列查看器和编辑器
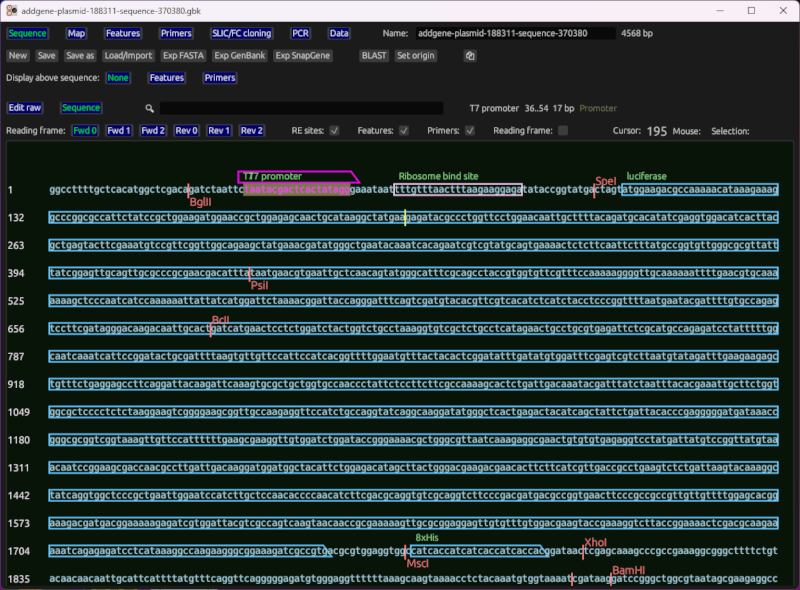
此功能显示感兴趣序列(从克隆或手动输入生成)以及根据其匹配位置叠加的引物。它还显示常见限制性内切酶的切割位点,以及从文件加载或由用户设置的特性。它包括编码区域的基本阅读框(ORF)查看。
它允许您以所见即所得的方式执行标准编辑操作。即,您可以通过单击序列来设置光标,使用箭头键移动光标,输入字母A、C、T和G来插入核苷酸等。
说明
- 双击以选择一个特性。
- Ctrl + F 以搜索序列。
- Ctrl+C 和 Ctrl+V 以复制和粘贴序列。
- 单击并拖动以选择文本。
- 使用键盘插入核苷酸、移动光标等。
- 当选择一个特征或引物时,将复制该选择,而不是选择本身。
- 特征和引物编辑表格可以在地图上方显示。
- 使用Esc键删除选择和光标。
环形图
正在编辑的质粒的环形序列图,其中显示特征、引物、限制酶位点和其他数据。
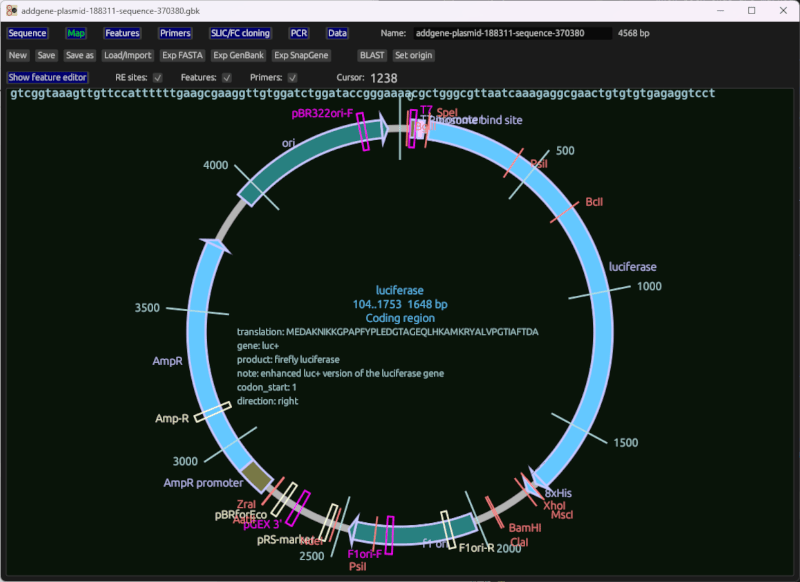
说明
- 点击选择一个特征
- 您可以使用滑块或顶部的相邻文本框更改特征范围。
- 特征编辑表可以在地图上方显示
特征和引物注释
可以创建、编辑和加载特征和引物。我们将引物定义为与序列中特定索引显式匹配的序列,并将引物定义为动态匹配的核苷酸序列。(TR)
限制酶和标签动态注释
自动识别和标记常见的限制酶位点,以及标签,如6x或8x HIS标签。
PCR参数生成
根据产物长度、引物熔解温度、聚合酶类型和其他参数生成PCR参数(例如,各步骤的温度和时间)。
与FASTA、GenBank和SnapGene互操作
可以读取和写入FASTA、GenBank和SnapGene .dna文件。FASTA文件仅存储序列数据,而GenBank、SnapGene和PlasCAD的自身格式存储序列数据、特征、引物和元数据。我们使用gb_io库来处理GenBank解析和写入。
要打开文件,您可以使用顶部的加载/导入按钮,或者将文件拖入窗口。
何时使用每种文件格式
如果您没有互操作性要求,可以使用PlasCAD的自身文件格式(.pcad扩展名),使用保存和加载按钮。这是一个二进制格式,与其他格式相比,文件大小更小(例如,4-10倍),因为每个核苷酸使用2位来存储,并且整体格式更紧凑。
GenBank和SnapGene是流行的格式,并且与其他软件提供互操作性。AddGene和其他资源提供这些格式的文件。
PlasCAD目前不能处理所有DNA格式的数据,也不能生成SnapGene可以打开的DNA文件。这意味着您可以打开DNA文件并获取大多数有用数据,但我们不建议在其中保存。考虑到这一点,GenBank是与其他软件兼容的最佳格式。
FASTA文件包含序列数据,因此使用此格式将导致特征和引物数据的丢失。
重要:此程序目前不能生成SnapGene原生格式文件,该文件可以被SnapGene打开。如果您正在寻找SnapGene双向兼容性,请在问题解决之前使用GenBank格式。
将文件与PlasCAD关联
如果您希望将文件类型(例如,原生的.pcad格式)与PlasCAD关联,您必须手动使用操作系统设置它,因为PlasCAD没有安装程序。例如在Windows上:右键单击文件 → 打开方式 → 选择另一个应用程序 → (选择您保存程序的位置) → 始终。
为何需要另一个质粒编辑器
我们相信,为科学家提供的工具越多,就越好。特别是,我的目标是制作一个快速、轻量级的程序,尽可能易于使用,而不牺牲功能。我们还添加了一些在其他地方看不到的功能,例如自动引物调整,以及为SLIC和FastCloning生成引物。
值得注意的是,该程序使用的原生文件格式更紧凑,包括DNA序列,其中每个核苷酸仅占用2位,而常见的格式占用8位。
近期计划
- 检查质粒中是否有毒蛋白质和各种错误形式
- 质粒问题中使用的QC引物。(例如,多重/部分结合位点)
- 识别二级结构、发夹等
- 特定应用的实用功能
- 更稳健的编辑功能
- 更好地处理重叠功能
使用的计算方法
所使用的引物熔点方法是基于SantaLucia & Hicks (2004)。它使用引物序列中相邻的每个核苷酸对来估计熵和焓值,这些值用于以下计算,其中$ΔH$是焓,$ΔS$是熵,$C_T$是引物的摩尔浓度
$$ (1000 * ΔH) / (ΔS + R \times ln(\frac{C_T}{4})) - 273.15 $$
计算还包括来自BioPython的盐校正,使用$K^+$、$Na^+$、$Mg^{2+}$和dntp浓度的浓度。这些由用户提供,或使用默认值启动。
我们计算以下类别的核苷酸重复
- 连续4次或更多次的单个核苷酸重复
- 连续4次或更多次的核苷酸对重复
- 在任何序列中重复出现3个或更多核苷酸序列
引物质量是一个模糊度量,它是从其他度量中计算出的加权平均值。它是我们的调优算法在调整引物长度时优化的数量。
依赖关系
~26–64MB
~1M SLoC