3个版本 (破坏性更新)
| 0.3.0 | 2024年6月20日 |
|---|---|
| 0.2.0 | 2022年6月13日 |
| 0.1.0 | 2021年11月4日 |
#124 in 生物学
105KB
621 行
![]()
![]()
![]()
![]()
从一个或两个比对文件计算成对的SNP距离矩阵
目录
动机
psdm的一个关键区别是两个比对文件之间的成对SNP距离。如果您已经为许多样本计算了SNP距离矩阵,并希望用一些新样本更新距离,而无需重新运行原始文件中所有样本的分析,这将特别有益。
另一个潜在用途是拥有两个具有相同样本但由不同技术生成的比对文件。例如,如果您从Illumina和Nanopore的SNP调用中生成了共识序列,并希望查看Illumina到Nanopore(跨技术)距离与跨技术距离相比如何。
尽管有这些动机,psdm仍然可以用来计算单个FASTA比对文件的“传统”成对SNP距离矩阵。
安装
cargo
![]()
先决条件:rust 工具链(最小版本v1.55.0)
$ cargo install psdm
conda
![]()
$ conda install psdm
预编译的二进制文件
tl;dr:运行以下片段将系统上的最新二进制文件下载到当前目录,并显示帮助菜单。
version="0.3.0"
OS=$(uname -s)
if [ "$OS" = "Linux" ]; then
triple="x86_64-unknown-linux-musl"
elif [ "$OS" = "Darwin" ]; then
triple="x86_64-apple-darwin"
else
echo "ERROR: $OS not a recognised operating system"
fi
if [ -n "$triple" ]; then
URL="https://github.com/mbhall88/psdm/releases/download/${version}/psdm-${version}-${triple}.tar.gz"
wget "$URL" -O - | tar -xzf -
./psdm --help
fi
这些二进制文件不需要您安装rust工具链。
目前,有两个预编译的二进制文件可用
- Linux内核
x86_64-unknown-linux-musl(在大多数我测试的Linux发行版上工作) - OSX内核
x86_64-apple-darwin(适用于任何2007年后的Mac)
使用wget下载这些二进制文件的一个示例
$ version="0.3.0"
$ URL="https://github.com/mbhall88/psdm/releases/download/${version}/psdm-${version}-x86_64-unknown-linux-musl.tar.gz"
$ wget "$URL" -O - | tar -xzf -
$ ./psdm --help
如果这些二进制文件在您的系统上不起作用,请提出问题,我将可能添加一些额外的目标三元组。
homebrew
先决条件:homebrew
使用homebrew-bio tap完成homebrew安装。
$ brew install brewsci/bio/psdm
容器
容器镜像托管在 quay.io。
singularity
先决条件:singularity
$ URI="docker://quay.io/mbhall88/psdm"
$ singularity exec "$URI" psdm --help
上述命令将使用最新版本。如果您想指定版本,请使用类似以下格式的 标签(或提交)。
$ VERSION="0.3.0"
$ URI="docker://quay.io/mbhall88/psdm:${VERSION}"
docker
先决条件:docker
$ docker pull quay.io/mbhall88/psdm
$ docker run quay.io/mbhall88/psdm psdm --help
您可以在 quay.io 仓库 中找到所有可用的标签。
本地
先决条件:rust 工具链
$ git clone https://github.com/mbhall88/psdm.git
$ cd psdm
$ cargo build --release
$ target/release/psdm --help
# if you want to check everything is working ok
$ cargo test
用法
快速
单个对齐文件
aln1.fa
>s1
ABCDEFGH
>s2
aBN-XFnH
>s0
AbCdEfG-
$ psdm aln1.fa
,s1,s2,s0
s1,0,1,0
s2,1,0,1
s0,0,1,0
两个对齐文件
aln2.fa.gz
>s2
xXNNfoo=
>s5 description
AB-DEFGG
$ psdm aln1.fa aln2.fa.gz
,s1,s2,s0
s2,6,5,5
s5,1,2,0
列名代表提供的 第一个 对齐文件。
完整
我想按标识符排序输出序列
$ psdm -s aln1.fa
,s0,s1,s2
s0,0,0,1
s1,0,0,1
s2,1,1,0
我想一个制表符分隔的(TSV)矩阵,而不是一个逗号分隔的(CSV)矩阵
$ psdm -d "\t" aln1.fa
s1 s2 s0
s1 0 1 0
s2 1 0 1
s0 0 1 0
核苷酸的字母大小写对我来说很重要 - 即,acgt 与 ACGT 不相同
$ psdm -c aln1.fa
,s1,s2,s0
s1,0,3,3
s2,3,0,5
s0,3,5,0
默认情况下,psdm 会忽略 N 和间隙(-)。但是,也许您还想忽略 X
$ psdm -e NX- aln1.fa
,s1,s2,s0
s1,0,0,0
s2,0,0,0
s0,0,0,0
或者,也许您不想忽略任何内容
$ psdm -e "" aln1.fa
,s1,s2,s0
s1,0,5,4
s2,5,0,8
s0,4,8,0
我很着急,请使用我所有的线程!
$ psdm -t 0 aln1.fa
请提供长格式输出而不是矩阵
$ psdm -l aln1.fa
s1,s1,0
s1,s2,1
s1,s0,0
s2,s1,1
s2,s2,0
s2,s0,1
s0,s1,0
s0,s2,1
s0,s0,0
我想知道成对比较的进度
$ psdm -P -t 8 aln.fa
[2024-06-20T02:50:32Z INFO psdm] Using 8 thread(s)
[2024-06-20T02:50:32Z INFO psdm] Loading first alignment file...
[2024-06-20T02:50:38Z INFO psdm] Loaded 200 sequences with length 4411532bp
[2024-06-20T02:50:38Z INFO psdm] Calculating 20100 pairwise distances...
Progress: 50.00% (10050 / 20100)
请将矩阵写入文件
$ psdm -o dists.csv aln1.fa
$ psdm --help
psdm 0.3.0
Michael Hall <michael@mbh.sh>
Compute a pairwise SNP distance matrix from one or two alignment(s)
USAGE:
psdm [OPTIONS] [ALIGNMENTS]...
ARGS:
<ALIGNMENTS>...
FASTA alignment file(s) to compute the pairwise distance for.
Providing two files will compute the distances for all sequences in one file against all
sequences from the other file - i.e., not between sequences in the same file. The first
file will be the column names, while the second is the row names. The alignment file(s)
can be compressed.
OPTIONS:
-c, --case-sensitive
Case matters - i.e., dist(a, A) = 1
-d, --delim <DELIMITER>
Delimiting character for the output table
[default: ,]
-e, --ignored-chars <IGNORED_CHARS>
String of characters to ignore - e.g., `-e N-` -> dist(A, N) = 0 and dist(A, -) = 0
Note, if using `--case-sensitive` the upper- and lower-case form of a character is
needed. To not ignore any characters, use `-e ''` or `-e ""`
[default: N-]
-h, --help
Print help information
-l, --long
Output as long-form ("melted") table
By default the output is a N x N or N x M table where N is the number of sequences in
the first alignment and M is the number of sequences in the (optional) second alignment.
-o, --output <OUTPUT>
Output file name [default: stdout]
-P, --progress
Show a progress bar
-q, --quiet
No logging (except progress info if `-P` is given)
-s, --sort
Sort the alignment(s) by ID
-t, --threads <THREADS>
Number of threads to use. Setting to 0 will use all available
[default: 1]
-V, --version
Print version information
基准测试
我们对 snp-dists(v0.8.2)进行基准测试。 snp-dists 是一个用于计算 SNP 距离矩阵的出色工具,也是 psdm 的灵感来源。
由于 snp-dists 不允许进行对齐文件间的比较,我们将基准测试内对齐模式。我们使用一个包含 151 个序列、长度为 4411532bp 的对齐文件。
基准测试时间使用 hyperfine 记录,每个工具/线程组合运行 10 次。
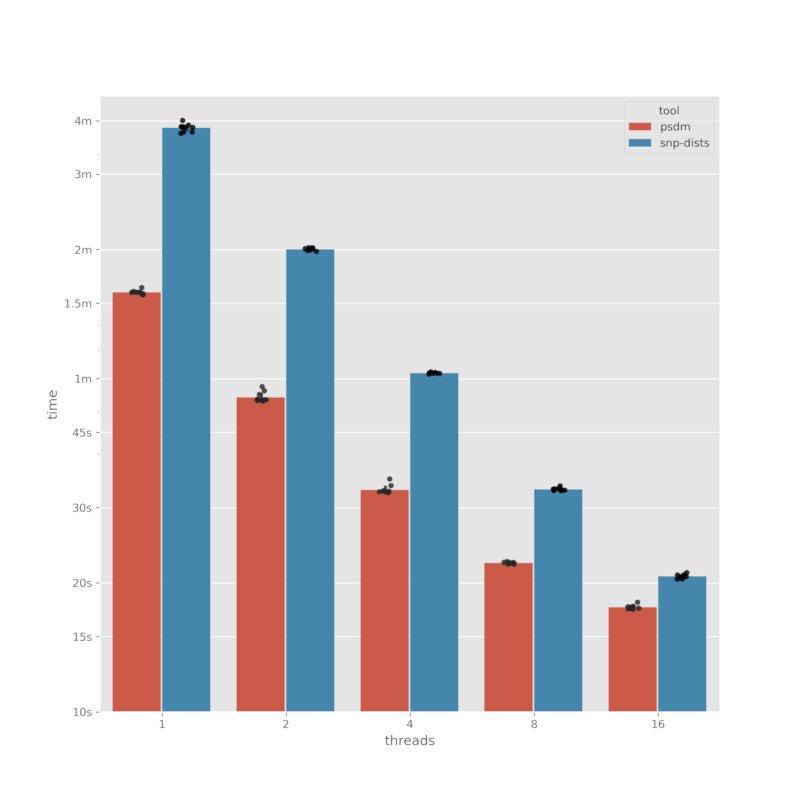
| 工具 | 线程 | 平均 | 标准差 | 最小值 | 中位数 | 最大值 |
|---|---|---|---|---|---|---|
| psdm | 1 | 95.4 | 1.0 | 94.2 | 95.3 | 97.9 |
| snp-dists | 1 | 231.0 | 4.8 | 224.2 | 231.8 | 240.4 |
| psdm | 2 | 54.3 | 1.5 | 53.2 | 53.6 | 57.5 |
| snp-dists | 2 | 120.3 | 0.8 | 118.9 | 120.3 | 121.3 |
| psdm | 4 | 33.0 | 0.8 | 32.5 | 32.7 | 35.0 |
| snp-dists | 4 | 61.8 | 0.2 | 61.5 | 61.8 | 62.2 |
| psdm | 8 | 22.3 | 0.1 | 22.1 | 22.3 | 22.4 |
| snp-dists | 8 | 33.1 | 0.3 | 32.8 | 33.1 | 33.7 |
| psdm | 16 | 17.6 | 0.2 | 17.4 | 17.5 | 18.0 |
| snp-dists | 16 | 20.7 | 0.2 | 20.4 | 20.7 | 21.1 |
贡献
欢迎贡献。要被接受,更改必须通过 CI 和覆盖率检查。这包括
- 代码格式化(
cargo fmt)。 - 没有编译器错误/警告。
cargo clippy --all-features --all-targets -- -D warnings - 测试代码覆盖率没有降低。
请还在 CHANGELOG 中添加贡献的简洁描述。
引用
psdm 已存档至 Zenodo。
@software{Hall2021psdm,
author = {Hall, Michael B.},
title = {{psdm: Compute a pairwise SNP distance matrix from
one or two alignment(s)}},
month = nov,
year = 2021,
publisher = {Zenodo},
version = {0.3.0},
doi = {10.5281/zenodo.5706784},
url = {https://doi.org/10.5281/zenodo.5706785}
}
依赖项
~12–20MB
~272K SLoC