4个版本
| 0.1.3 | 2022年3月22日 |
|---|---|
| 0.1.2 | 2021年12月29日 |
| 0.1.1 | 2021年12月29日 |
| 0.1.0 | 2021年12月28日 |
#144 in 生物学
105KB
1K SLoC
guide-counter
![]()
![]()
![]()
一种更好的、更快的方法来统计CRISPR筛选中的指南。
概述
guide-counter是一个工具,用于处理CRISPR筛选实验的FASTQ文件,以生成每个样本的指南计数矩阵。它可以作为比mageck count更快、更准确的替代品。默认情况下,guide-counter将在与预期指南相比有0或1个错配的读取中查找指南序列,但也可以以精确匹配模式运行。
为什么选择guide-counter?
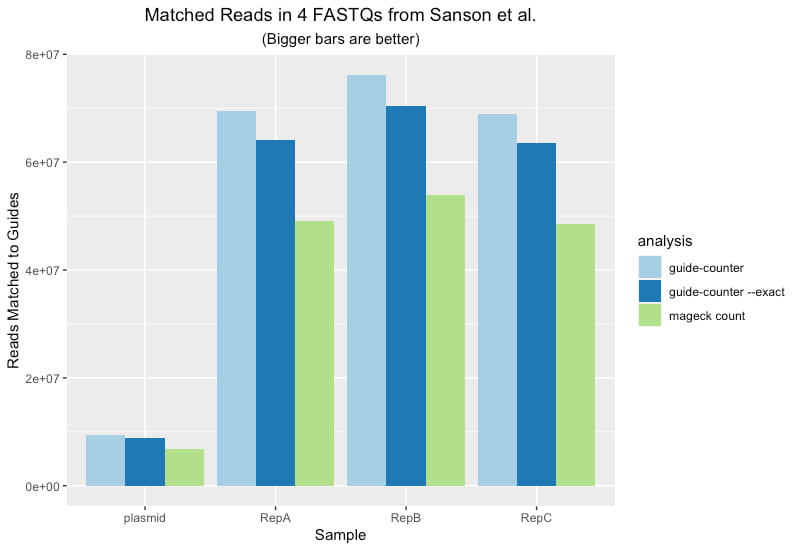
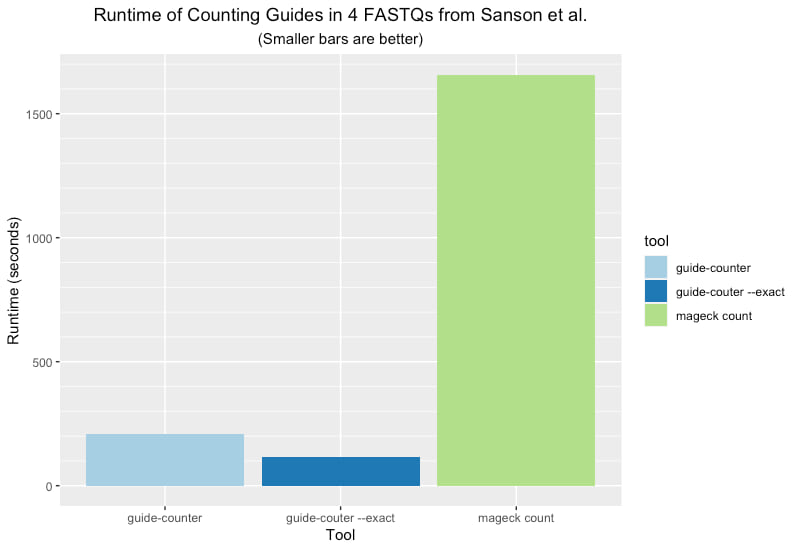
如果您有分析CRISPR筛选的经验,您几乎肯定尝试过mageck。它被广泛使用,高度引用,并且通常表现良好。然而,出人意料的是,mageck count既慢又漏掉了相当数量的数据。
例如,我们将来自Sanson等人论文中的数据通过这两个工具运行。数据集包括
| 样本 | 读取 | gziped FASTQ大小 |
|---|---|---|
| 质粒 | 9,821,128 | 377M |
| RepA | 76,471,324 | 2.3G |
| RepB | 85,301,059 | 2.5G |
| RepC | 75,356,900 | 2.2G |
以下图表显示了每个样本通过三种不同分析恢复的数据量
以下图表显示了使用单个CPU核心/线程在配备Intel Core i9的MacBook Pro笔记本电脑上执行的三种分析运行时间
安装
可以使用conda进行安装
conda install -c bioconda guide-counter
或者如果有安装,可以使用cargo
cargo install guide-counter
示例工作流程
以下显示了在Sanson等人2018年论文数据上运行guide-counter然后运行mageck test的示例
guide-counter count \
--input plasmid.fq.gz RepA.fq.gz RepB.fq.gz RepC.fq.gz \
--control-pattern control \
--essential-genes metadata/training_essentials.txt \
--nonessential-genes metadata/training_nonessential.txt \
--library metadata/broadgpp-brunello-library-corrected.txt.gz \
--output sanson
mageck test \
--count-table sanson.counts.txt \
--control-id plasmid \
--treatment-id RepA,RepB,RepC \
--norm-method median \
--output-prefix sanson.test
输入
以下重现了guide-counter count的完整用法;本节详细描述了一些关键输入
| 输入选项 | 必需 | 描述 |
|---|---|---|
--input |
是 | 每个样本一个FASTQ文件。文件可以是gzip压缩或未压缩的。 |
--samples |
否 | 样本名称,与FASTQ文件位置匹配。如果未提供,则输入文件名减去任何`.[fq |
--essential-genes |
否 | 一个可选的已知必需基因文件。可以是gzip压缩或未压缩的。可以是每行一个基因名称,也可以是第一列包含基因的制表符分隔的。如果提供,则会将引导标记为针对匹配基因的必需,并计算必需基因的引导平均覆盖率。 |
--nonessential-genes |
否 | 一个可选的已知非必需基因文件。可以是gzip压缩或未压缩的。可以是每行一个基因名称,也可以是第一列包含基因的制表符分隔的。如果提供,则会将引导标记为针对匹配基因的非必需,并计算非必需基因的引导平均覆盖率。 |
--控制-guides |
否 | 一个可选的控制引导ID文件。可以是gzip压缩或未压缩的。可以是每行一个引导ID,也可以是第一列包含引导ID的制表符分隔的数据。如果提供,则匹配的引导将被标记为控制,并计算控制引导的平均覆盖率。可以单独使用或与--control-pattern一起使用。 |
--控制-pattern |
否 | 一个可选的正则表达式,应用于引导ID和基因名称(不区分大小写),并且当找到匹配项时,将引导标记为控制。例如,--control-pattern control对于许多人类库来说效果很好。 |
输出
输出文件生成
{output}.counts.txt- 一个标准计数矩阵,包含引导ID和基因的列,然后是每个样本的原始/未归一化引导计数列。{output}.-extended-counts.txt- 计数矩阵的扩展版本,包含一个guide_type列,其中每个引导将根据提供的基因列表和控制信息确定一个[Essential, Nonessential, Control, Other]。{output}.stats.txt- 一个计算统计文件,每行一个输入样本/FASTQ。
统计文件中的列
| 列 | 描述 |
|---|---|
| file | 用于生成统计的输入FASTQ文件的路径。 |
| label | 提供给样本的标签或样本名称。 |
| total_guides | 引导库中的引导总数(不依赖于样本)。 |
| total_reads | 输入FASTQ文件中的总读取数。 |
| mapped_reads | 可以映射到引导的读取数。 |
| frac_mapped | 可以映射到引导的读取比例(0-1)。 |
| mean_reads_per_guide | 库中每个引导的映射读取的平均数。 |
| mean_reads_essential | 映射到必需基因引导的平均读取数。 |
| mean_reads_nonessential | 映射到非必需基因引导的平均读取数。 |
| mean_reads_control | 映射到控制引导的平均读取数。 |
| mean_reads_other | 映射到其他引导(非必需、非必需或控制标记的引导)的平均读取数量。 |
| zero_read_guides |
使用方法
使用方法:guide-counter count
guide-counter-count
Counts the guides observed in a CRISPR screen, starting from one or more FASTQs. FASTQs are one per
sample and currently only single-end FASTQ inputs are supported.
A set of sample IDs may be provided using `--samples id1 id2 ..`. If provided it must have the same
number of values as input FASTQs. If not provided the FASTQ names are used minus any fastq/fq/gz
suffixes.
Automatically determines the range of valid offsets within the sequencing reads where the guide
sequences are located, independently for each FASTQ input. The first `offset-sample-size` reads
from each FASTQ are examined to determine the offsets at which guides are found. When processing the
full FASTQ, checks only those offsets that accounted for at least `offset-min-fraction` of the first
`offset-sample-size` reads.
Matching by default allows for one mismatch (and no indels) between the read sub-sequence and the
expected guide sequences. Exact matching may be enabled by specifying the `--exact-match` option.
Two output files are generated. The first is named `{output}.counts.txt` and contains columns for
the guide id, the gene targeted by the guide and one count column per input FASTQ with raw/un-
normalized counts. The second is named `{output}.stats.txt` and contains basic QC statistics per
input FASTQ on the matching process.
USAGE:
guide-counter count [OPTIONS] --input <INPUT>... --library <LIBRARY> --output <OUTPUT>
OPTIONS:
-c, --control-guides <CONTROL_GUIDES>
Optional path to file with list control guide IDs. IDs should appear one per line and
are case sensitive
-C, --control-pattern <CONTROL_PATTERN>
Optional regular expression pattern used to ID control guides. Pattern is matched, case
insensitive, to guide IDs and Gene names
-e, --essential-genes <ESSENTIAL_GENES>
Optional path to file with list of essential genes. Gene names should appear one per
line and are case sensitive
-f, --offset-min-fraction <OFFSET_MIN_FRACTION>
After sampling the first `offset_sample_size` reads, use offsets that
[default: 0.005]
-h, --help
Print help information
-i, --input <INPUT>...
Input fastq file(s)
-l, --library <LIBRARY>
Path to the guide library metadata. May be a tab- or comma-separated file. Must have a
header line, and the first three fields must be (in order): i) the ID of the guide, ii)
the base sequence of the guide, iii) the gene the guide targets
-n, --nonessential-genes <NONESSENTIAL_GENES>
Optional path to file with list of nonessential genes. Gene names should appear one per
line and are case sensitive
-N, --offset-sample-size <OFFSET_SAMPLE_SIZE>
The number of reads to be examined when determining the offsets at which guides may be
found in the input reads
[default: 100000]
-o, --output <OUTPUT>
Path prefix to use for all output files
-s, --samples <SAMPLES>...
Sample names corresponding to the input fastqs. If provided must be the same length as
input. Otherwise will be inferred from input file names
-x, --exact-match
Perform exact matching only, don't allow mismatches between reads and guides
依赖关系
~10–20MB
~258K SLoC