2个稳定版本
| 1.1.0 | 2021年10月8日 |
|---|---|
| 1.0.1 | 2021年8月25日 |
#142 in 生物学
2MB
2.5K SLoC
FragGeneScanRs
安装
从版本
下载适用于您平台的最新版本构建,并将其解压缩到路径中某个位置。
从源码
FragGeneScanRs是用Rust编写的,因此首先请参阅他们的安装说明。之后,您可以使用cargo install frag_gene_scan_rs从crates.io安装crate,或者您可以从这里安装。克隆此存储库或下载最新版本的源代码。在此目录下,运行cargo install --path .进行安装。安装过程可能会提示您将目录添加到路径中,以便您可以轻松执行它。
用法
您可以使用FragGeneScanRs与FragGeneScan的简短选项一起使用,但它还提供长格式选项和一些附加选项。它默认从标准输入读取并写入标准输出,允许在您只需要预测的蛋白质的情况下进行更短的调用。
# get predictions for 454 pyrosequencing reads with about 1% error rate
FragGeneScanRs -t 454_10 < example/NC_000913-454.fna > example/NC_000913-454.faa
# get predictions for complete reads
FragGeneScanRs -t complete -w 1 < example/NC_000913.fna > example/NC_000913.faa
向下兼容模式
FragGeneScanRs -s seq_file_name -o output_file_name -w [0 or 1] -t train_file_name -p num_threads
其中
-
seq_file_name是需要进行基因预测的DNA序列的FASTA文件的绝对路径 -
output_file_name是三个输出文件的绝对路径和前缀。将分别创建扩展名为.out、.faa和.ffn的文件,分别包含基因预测元数据、预测基因的蛋白质翻译和预测基因的DNA序列。 -
0或1用于短序列读数或完整的基因组序列。 -
train_file_name用于选择以下类型之一的训练文件:complete用于完整的基因组序列或无测序错误的短序列读数sanger_5用于约0.5%错误率的Sanger测序读取sanger_10用于约1%错误率的Sanger测序读取454_5用于约0.5%错误率的454焦磷酸测序读取454_10用于约1%错误率的454焦磷酸测序读取454_30用于约3%错误率的454焦磷酸测序读取illumina_5用于约0.5%错误率的Illumina测序读取illumina_10用于约1%错误率的Illumina测序读取
相应的文件应位于工作目录下的子目录
train中。其他文件可以在此处添加和选择。 -
num_threads是要使用的线程数。默认为1。
其他选项
-
-m meta_file、-n nucleotide_file、-a aa_file和-g gff_file可用于将输出写入特定文件,而不是让程序创建具有预定扩展名的文件名。这些选项优先于-o选项。 -
省略
-o选项或使用名称stdout将导致 FragGeneScanRs 只将预测的蛋白质写入标准输出。其他文件仍可以使用上述特定选项请求。 -
省略
-s选项将导致 FragGeneScanRs 从标准输入读取序列。 -
-r train_file_dir允许显式指定包含训练文件的目录路径,因此您可以在系统上的任何位置执行命令。 -
选项
-u可用于在使用多线程时提供一些额外的速度和减少内存。输出将不再按输入顺序排列(如FGS和FGS+所示)。
当运行 FragGeneScanRs --help 时,将打印出完整的选项列表。
执行时间(版本1.0.0)
使用 meta/benchmark.sh 脚本在16核心Intel(R) Xeon(R) CPU E5-2650 v2 @ 2.60GHz和195GB RAM的机器上进行了基准测试。使用的数据集是 FragGeneScan 提供的示例数据集。下表显示了5次运行的平均执行时间。详细结果可以在 meta/benchmark.csv 中找到。对于短读(80bp),FragGeneScanRs 比FragGeneScan快约22倍,比FragGeneScanPlus快1.2倍。对于长读(1328bp)和完整基因组(大肠杆菌K-12亚种MG1655,4639675bp),FragGeneScanRs 分别比FragGeneScan快4.3倍和2.2倍,比FGS+快1.6倍和233.6倍。
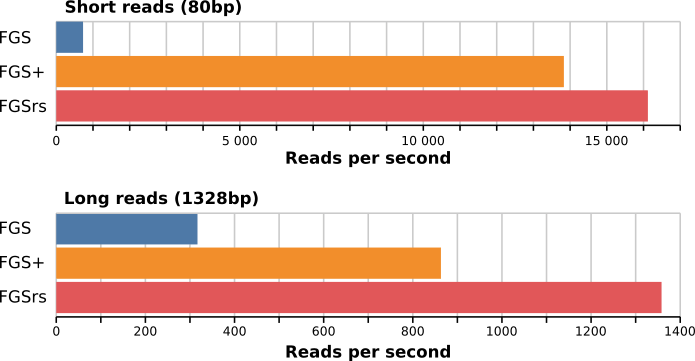
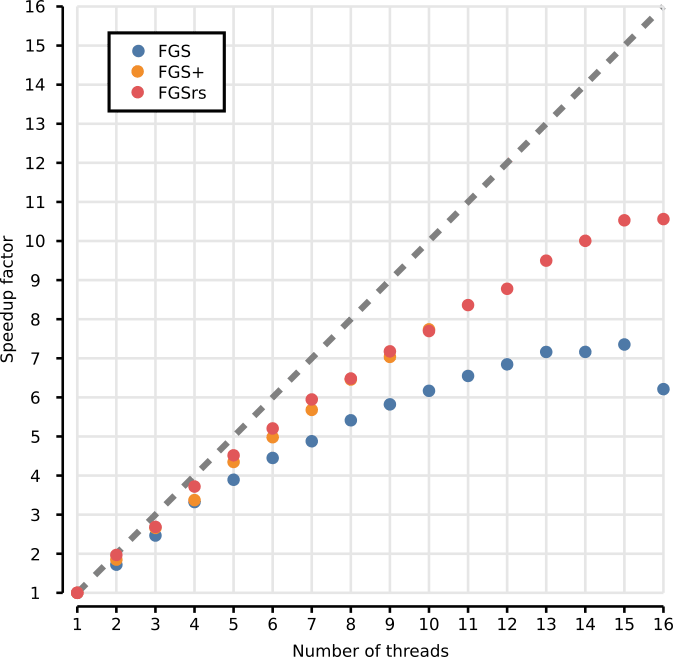
| 短读 | 1 线程 | 2 线程 | 4 线程 | 8 线程 | 16 线程 |
|---|---|---|---|---|---|
| FragGeneScan | 731 r/s | 1257 r/s | 2158 r/s | 3408 r/s | 3371 r/s |
| FragGeneScanPlus | 13830 r/s | 23997 r/s | 37882 r/s | 54610 r/s | / |
| FragGeneScanRs | 16119 r/s | 29326 r/s | 48593 r/s | 73965 r/s | 99885 r/s |
| 长读 | 1 线程 | 2 线程 | 4 线程 | 8 线程 | 16 线程 |
|---|---|---|---|---|---|
| FragGeneScan | 317 r/s | 545 r/s | 1053 r/s | 1715 r/s | 1968 r/s |
| FragGeneScanPlus | 863 r/s | 1596 r/s | 2910 r/s | 5573 r/s | / |
| FragGeneScanRs | 1358 r/s | 2674 r/s | 5051 r/s | 8803 r/s | 14343 r/s |
| 完整基因组 | 1 线程 |
|---|---|
| FragGeneScan | 6.668 秒 |
| FragGeneScanPlus | 712.265 秒 |
| FragGeneScanRs | 3.049 秒 |
用于这些基准测试的命令和参数是
./FragGeneScan -t 454_10 -s example/NC_000913-454.fna -o example/NC_000913-454 -w 0
./FGS+ -t 454_10 -s example/NC_000913-454.fna -o example/NC_000913-454 -w 0
./FragGeneScanRs -t 454_10 -s example/NC_000913-454.fna -o example/NC_000913-454 -w 0
./FragGeneScan -t complete -s example/contigs.fna -o example/contigs -w 1
./FGS+ -t complete -s example/contigs.fna -o example/contigs -w 1
./FragGeneScanRs -t complete -s example/contigs.fna -o example/contigs -w 1
./FragGeneScan -t complete -s example/NC_000913.fna -o example/NC_000913 -w 1
./FGS+ -t complete -s example/NC_000913.fna -o example/NC_000913 -w 1
./FragGeneScanRs -t complete -s example/NC_000913.fna -o example/NC_000913 -w 1
默认情况下,FragGeneScanPlus只输出预测的基因,不输出元数据和DNA文件。以下是在FragGeneScanRs也没有生成这些文件时的测量结果。
| 短读 | 1 线程 | 2 线程 | 4 线程 | 8 线程 |
|---|---|---|---|---|
| FragGeneScanPlus | 13765 次/秒 | 24500 次/秒 | 39548 次/秒 | 57147 次/秒 |
| FragGeneScanRs | 16815 次/秒 | 28784 次/秒 | 50157 次/秒 | 75397 次/秒 |
这里使用的命令是
./FGS+ -t 454_10 -s example/NC_000913-454.fna -o stdout -w 0 > /dev/null
./FragGeneScanRs -t 454_10 -s example/NC_000913-454.fna -o stdout -w 0 > /dev/null
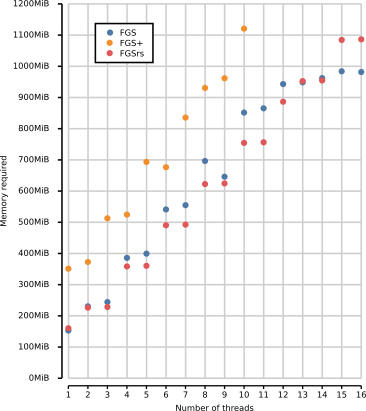
内存使用(版本1.0.0)
下方的图表显示了FGS、FGS+和FGSrs在长读长(1328 bp)上的多线程执行内存占用。总内存占用(堆、栈和内存映射文件I/O)使用Valgrind的Massif堆分析器测量,并带有--pages-as-heap选项。在10个线程以上时,FGS+的竞争条件会一直阻止执行。FGS和FGSrs生成DNA序列、蛋白质翻译和元数据,而FGS+仅生成蛋白质翻译,因为生成其他输出时软件会崩溃。FGS和FGS+报告基因预测的顺序与默认的顺序报告不同,FGSrs使用了默认的顺序报告。
依赖项
约5MB
约93K SLoC