1 个不稳定版本
| 0.1.0 | 2024 年 4 月 27 日 |
|---|
#95 在 生物学 中
145KB
1K SLoC
Ambigviz
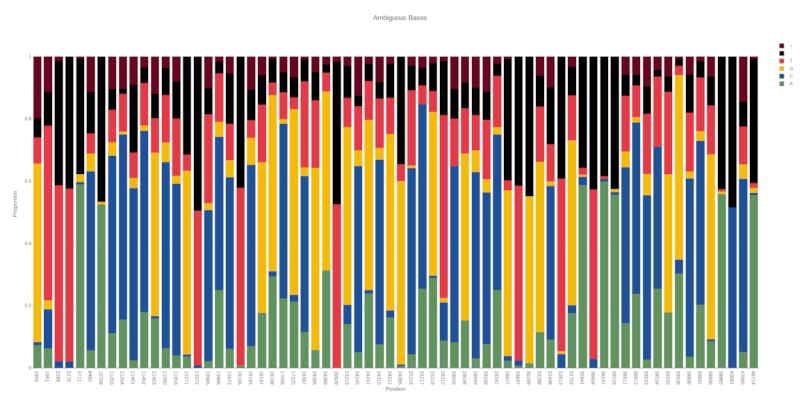
Ambigviz 是一个用于快速扫描和可视化 BAM 文件中给定位置的模糊/混合碱基的工具。最初,它被编写来检查 SARS-CoV-2 样本中的宿主内多样性和共感染,但它可以用于任何 BAM 文件(并得益于 Rust 的良好扩展性)。它默认使用严格的过滤选项,以避免测序伪迹、污染/测序错误。其目的是快速生成“适合展示”的图表,以便进行进一步的沟通和讨论。
它提供了一个简单的命令行界面,并且仅需一个 BAM 文件即可。
安装
Cargo
需要 cargo
cargo install ambigviz
从源码构建
安装 Rust 工具链
有关安装信息,请参阅 Rust 文档: 文档
克隆仓库
git clone https://github.com/Sam-Sims/ambigviz
构建并添加到路径
cd ambigviz
cargo build --release
export PATH=$PATH:$(pwd)/target/release
所有可执行文件都将位于 ambigviz/target/release 目录中。
使用方法
基本用法
ambigviz ambig <path_to_bam> <region> [options]
至少需要 BAM 文件。Ambigviz 将在同一目录中查找以 .bai 模式匹配的 [input].bai 的 .bai 文件 - 如果找不到,Ambigviz 将尝试为您索引 BAM 文件。如果失败,您可以使用 samtools 手动索引 BAM 文件。
默认情况下,如果没有提供区域,将扫描整个 BAM 文件以查找模糊碱基。结合默认的 20% 模糊阈值,这为快速扫描 BAM 文件中的测序错误、污染或共感染以及标记感兴趣的区域以进行进一步调查提供了一种简单的方法。
区域遵循 samtools 格式: chr:start-end,并且所有位置都是基于 1 的。
--bed 选项允许您将识别的模糊位置输出到 bed 文件。
您还可以使用深度命令绘制 BAM 文件的深度。
ambigviz depth <path_to_bam> <region> [options]
选项
输出
-o, --输出<输出>
此选项将设置输出文件名。默认为输出染色体名称。
阈值
-t, --阈值<阈值>
此选项将设置给定位置模糊碱基比例的阈值。超过此阈值的任何位置都将绘制。最大值为 0.5(50%)。默认为 0.2(20%)。
插入/删除
--no-indel
默认情况下,插入和缺失(indels)被包含在模糊碱基计数中。此选项将排除它们。这对于像ONT这样的噪声数据很有用。
深度
-D, --深度<深度>
此选项将设置一个位置被包含在图中的最小总深度。默认值为100。
次要深度。
-d, --次要-深度<次要-深度>
此选项将设置一个位置被包含在图中的次要等位基因的最小深度。默认值为20。
碱基质量
-q, --碱基-质量<碱基-质量>
此选项将设置一个碱基被包含在图中的最小碱基质量。默认值为20。
比对质量
-Q, --比对-质量<比对-质量>
此选项将设置一个读取被包含在图中的最小比对质量。默认值为60。
链偏斜
-s, --链-偏差<链-偏差>
此选项将设置一个位置被包含在图中的链偏差阈值。默认值为0.1(10%)。这意味着至少有10%的深度必须来自任一链。例如,如果某个位置有80个A读取和20个C读取,那么为了将位置标记为模糊并包含在图中,C中至少必须有来自每个链的2个读取。最大值为0.5,要求两个链的深度相等。设置为0将禁用此过滤器。
Bed文件
--bed
此选项将输出已识别的模糊位置到bed文件中。默认情况下不输出。
标签
--no-labels
默认情况下,每个位置每个碱基的比例作为文本注释包含在内。此选项将排除它们。
依赖关系
~21-33MB
~513K SLoC