30 个版本
| 0.7.7 | 2024 年 7 月 19 日 |
|---|---|
| 0.7.5 | 2023 年 7 月 28 日 |
| 0.7.1 | 2022 年 6 月 14 日 |
| 0.6.1 |
|
| 0.4.1 | 2019 年 9 月 9 日 |
#18 in 生物学
441 每月下载量
在 nwr 中使用
4MB
8K SLoC
intspan
![]()
![]()
![]()
安装
当前版本:0.7.7
cargo install intspan
cargo install --force --path .
# or
brew install intspan
# build under WSL 2
export CARGO_TARGET_DIR=/tmp
cargo build
cargo run --bin fasr help
# local docs
cargo doc --open
概念
范围
例如,S288c.rg。此格式呈现的信息与 BED 等格式非常相似。
我选择这种格式是因为其紧凑性、可读性和可嵌入其他制表符分隔文件的能力。
I:1-100
I(+):90-150
S288c.I(-):190-200
II:21294-22075
II:23537-24097
以下是一个 Range 对象的架构。
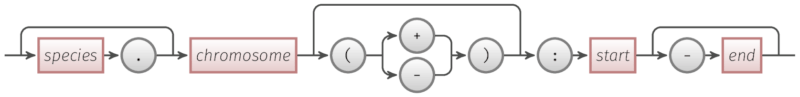
简单规则
chromosome和start是必需的species、strand和end是可选的.用于分隔species和chromosomestrand是+或-之一,并用圆括号括起来:用于分隔名称和数字-用于分隔start和end- 关于
speciesspecies应该是字母数字的,没有空格,一个例外字符是/。species是一个标识符,你也可以将其视为一个菌株名称、一个组装或其它。
species.chromosome(strand):start-end
--------^^^^^^^^^^--------^^^^^^----
在这个工具集中,rgr 用于操作 .rg 和 .tsv 文件中的范围。
IntSpans
IntSpan 表示整数集的表示方法,以包含范围的数量,例如 1-10,19,45-48。
以下图显示了 IntSpan 对象的架构。跳行在基线之上;循环线在基线之下。
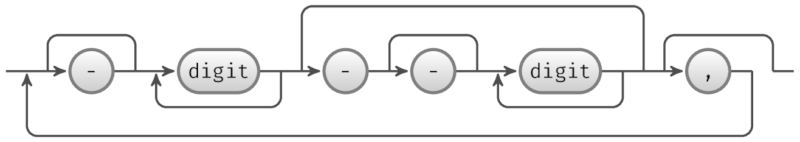
此外,AlignDB::IntSpan 和 jintspan 分别是 Perl 和 Java 中 IntSpan 对象的实现。
运行列表 - 存储在 JSON 中的染色体上的 IntSpan
我们经常需要处理具有相同属性的许多基因组区间,例如,一个基因的所有外显子,一个基因家族的所有启动子,一个基因组中的所有重复序列等。
现有的格式,如 bedGraph,可以部分处理这种情况,但往往面临直观性、性能等问题。同时,能够处理此类专有格式的工具数量非常有限。
将 IntSpan 保存到 JSON 文件是该工具集的解决方案,其中 spanr 处理这项工作。
- 单个:
repeat.json
{
"I": "-",
"II": "327069-327703",
"III": "-",
"IV": "512988-513590,757572-759779,802895-805654,981142-987119,1017673-1018183,1175134-1175738,1307621-1308556,1504223-1504728",
"IX": "-",
"V": "354135-354917",
"VI": "-",
"VII": "778784-779515,878539-879235",
"VIII": "116405-117059,133581-134226",
"X": "366757-367499,712641-713226",
"XI": "162831-163399",
"XII": "64067-65208,91960-92481,451418-455181,455933-457732,460517-464318,465070-466869,489753-490545,817840-818474",
"XIII": "609100-609861",
"XIV": "-",
"XV": "437522-438484",
"XVI": "560481-561065"
}
- 多个:
Atha.json
{
"AT1G01010.1": {
"1": "3631-3913,3996-4276,4486-4605,4706-5095,5174-5326,5439-5899"
},
"AT1G01020.1": {
"1": "5928-6263,6437-7069,7157-7232,7384-7450,7564-7649,7762-7835,7942-7987,8236-8325,8417-8464,8571-8737"
},
"AT1G01020.2": {
"1": "6790-7069,7157-7450,7564-7649,7762-7835,7942-7987,8236-8325,8417-8464,8571-8737"
},
"AT2G01008.1": {
"2": "1025-1272,1458-1510,1873-2810,3706-5513,5782-5945"
},
"AT2G01021.1": {
"2": "6571-6672"
}
}
chr.sizes:S288c.chr.sizes
范围链接
链接类型
-
双边链接
I(+):13063-17220 I(-):215091-219225 I(+):139501-141431 XII(+):95564-97485 -
带有命中链的双边链接
I(+):13327-17227 I(+):215084-218967 - I(+):139501-141431 XII(+):95564-97485 + -
多边链接
II(+):186984-190356 IX(+):12652-16010 X(+):12635-15993
概要
rgr help
`rgr` operates ranges in .rg and .tsv files
Usage: rgr [COMMAND]
Commands:
count Count each range overlapping with other range files
field Create/append ranges from fields
merge Merge overlapped ranges via overlapping graph
prop Proportion of the ranges intersecting a runlist file
replace Replace fields in .tsv file
runlist Filter .rg and .tsv files by comparison with a runlist file
sort Sort .rg and .tsv files by a range field
help Print this message or the help of the given subcommand(s)
Options:
-h, --help Print help
-V, --version Print version
* Field numbers in the TSV file start at 1
spanr help
`spanr` operates chromosome IntSpan files
Usage: spanr [COMMAND]
Commands:
genome Convert chr.size to runlists
some Extract some records from a runlist json file
merge Merge runlist json files
split Split a runlist json file
stat Coverage on chromosomes for runlists
statop Coverage on chromosomes for one JSON crossed another
combine Combine multiple sets of runlists in a json file
compare Compare one JSON file against others
span Operate spans in a JSON file
cover Output covers on chromosomes
coverage Output minimum or detailed depth of coverage on chromosomes
gff Convert gff3 to covers on chromosomes
convert Convert runlist file to ranges file
help Print this message or the help of the given subcommand(s)
Options:
-h, --help Print help
-V, --version Print version
fasr help
`fasr` operates block fasta files
Usage: fasr [COMMAND]
Commands:
axt2fas Convert axt to block fasta
check Check genome locations in block fasta headers
concat Concatenate sequence pieces of the same species
consensus Generate consensus sequences by POA
cover Output covers on chromosomes
create Create block fasta files from links of ranges
filter Filter blocks, and can also be used as a formatter
join Join multiple block fasta files by a common target
link Output bi/multi-lateral range links
maf2fas Convert maf to block fasta
name Output all species names
pl-p2m Pipeline - pairwise alignments to multiple alignments
refine Realign files with external programs and trim unwanted regions
replace Concatenate sequence pieces of the same species
separate Separate block fasta files by species
slice Extract alignment slices
split Split block fasta files to per-alignment/chromosome fasta files
stat Extract a subset of species
subset Extract a subset of species
variation List variations (substitutions/indels)
help Print this message or the help of the given subcommand(s)
Options:
-h, --help Print help
-V, --version Print version
linkr help
`linkr` operates ranges on chromosomes and links of ranges
Usage: linkr [COMMAND]
Commands:
circos Convert links to circos links or highlights
sort Sort links and ranges within links
filter Filter links by numbers of ranges or length differences
clean Replace ranges within links, incorporate hit strands and remove nested links
connect Connect bilateral links into multilateral ones
help Print this message or the help of the given subcommand(s)
Options:
-h, --help Print help
-V, --version Print version
示例
spanr
spanr genome tests/spanr/S288c.chr.sizes
spanr genome tests/spanr/S288c.chr.sizes |
spanr stat tests/spanr/S288c.chr.sizes stdin --all
spanr some tests/spanr/Atha.json tests/spanr/Atha.list
spanr merge tests/spanr/I.json tests/spanr/II.json
spanr merge tests/spanr/I.json tests/spanr/II.other.json --all
spanr cover tests/spanr/S288c.rg
spanr cover tests/spanr/dazzname.rg
spanr coverage tests/spanr/S288c.rg -m 2
spanr coverage tests/spanr/S288c.rg -d
spanr gff tests/spanr/NC_007942.gff --tag tRNA
spanr span --op cover tests/spanr/brca2.json
spanr combine tests/spanr/Atha.json
spanr compare \
--op intersect \
tests/spanr/intergenic.json \
tests/spanr/repeat.json
spanr compare \
--op intersect \
tests/spanr/I.II.json \
tests/spanr/I.json \
tests/spanr/II.json
spanr split tests/spanr/I.II.json
spanr stat tests/spanr/S288c.chr.sizes tests/spanr/intergenic.json
spanr stat tests/spanr/S288c.chr.sizes tests/spanr/I.II.json
spanr stat tests/spanr/Atha.chr.sizes tests/spanr/Atha.json
spanr statop \
--op intersect \
tests/spanr/S288c.chr.sizes \
tests/spanr/intergenic.json \
tests/spanr/repeat.json
spanr statop \
--op intersect --all\
tests/spanr/Atha.chr.sizes \
tests/spanr/Atha.json \
tests/spanr/paralog.json
spanr convert tests/spanr/repeat.json tests/spanr/intergenic.json |
spanr cover stdin |
spanr stat tests/spanr/S288c.chr.sizes stdin --all
spanr merge tests/spanr/repeat.json tests/spanr/intergenic.json |
spanr combine stdin |
spanr stat tests/spanr/S288c.chr.sizes stdin --all
rgr
rgr field tests/Atha/chr.sizes --chr 1 --start 2 -a -s
rgr field tests/spanr/NC_007942.gff -H --chr 1 --start 4 --end 5 --strand 7 --eq 3:tRNA --ne '7:+'
rgr field tests/rgr/ctg.tsv --chr 2 --start 3 --end 4 -H -f 6,1 > tests/rgr/ctg.range.tsv
rgr sort tests/rgr/S288c.rg
rgr sort tests/rgr/ctg.range.tsv -H -f 3
# ctg:I:1 is treated as a range
rgr sort tests/rgr/S288c.rg tests/rgr/ctg.range.tsv
rgr count tests/rgr/S288c.rg tests/rgr/S288c.rg
rgr count tests/rgr/ctg.range.tsv tests/rgr/S288c.rg -H -f 3
rgr runlist tests/rgr/intergenic.json tests/rgr/S288c.rg --op overlap
rgr runlist tests/rgr/intergenic.json tests/rgr/ctg.range.tsv --op non-overlap -H -f 3
rgr prop tests/rgr/intergenic.json tests/rgr/S288c.rg
rgr prop tests/rgr/intergenic.json tests/rgr/ctg.range.tsv -H -f 3 --prefix --full
rgr merge tests/rgr/II.links.tsv -c 0.95
rgr replace tests/rgr/1_4.ovlp.tsv tests/rgr/1_4.replace.tsv
rgr replace tests/rgr/1_4.ovlp.tsv tests/rgr/1_4.replace.tsv -r
# ctg_2_1_.gc.tsv isn't sorted,
cat tests/rgr/ctg_2_1_.gc.tsv | rgr sort stdin | cargo run --bin rgr pl-2rmp stdin > /dev/null
cat tests/rgr/II.links.tsv | cargo run --bin rgr pl-2rmp stdin
linkr
linkr sort tests/linkr/II.links.tsv -o tests/linkr/II.sort.tsv
rgr merge tests/linkr/II.links.tsv -v
linkr clean tests/linkr/II.sort.tsv
linkr clean tests/linkr/II.sort.tsv --bundle 500
linkr clean tests/linkr/II.sort.tsv -r tests/linkr/II.merge.tsv
linkr connect tests/linkr/II.clean.tsv -v
linkr filter tests/linkr/II.connect.tsv -n 2
linkr filter tests/linkr/II.connect.tsv -n 3 -r 0.99
linkr circos tests/linkr/II.connect.tsv
linkr circos --highlight tests/linkr/II.connect.tsv
步骤
sort
|
v
clean -> merge
| /
| /
v
clean
|
V
connect
|
v
filter
S288c
linkr sort tests/S288c/links.lastz.tsv tests/S288c/links.blast.tsv \
-o tests/S288c/sort.tsv
linkr clean tests/S288c/sort.tsv \
-o tests/S288c/sort.clean.tsv
rgr merge tests/S288c/sort.clean.tsv -c 0.95 \
-o tests/S288c/merge.tsv
linkr clean tests/S288c/sort.clean.tsv -r tests/S288c/merge.tsv --bundle 500 \
-o tests/S288c/clean.tsv
linkr connect tests/S288c/clean.tsv -r 0.8 \
-o tests/S288c/connect.tsv
linkr filter tests/S288c/connect.tsv -r 0.8 \
-o tests/S288c/filter.tsv
wc -l tests/S288c/*.tsv
# 229 tests/S288c/clean.tsv
# 148 tests/S288c/connect.tsv
# 148 tests/S288c/filter.tsv
# 566 tests/S288c/links.blast.tsv
# 346 tests/S288c/links.lastz.tsv
# 74 tests/S288c/merge.tsv
# 282 tests/S288c/sort.clean.tsv
# 626 tests/S288c/sort.tsv
cat tests/S288c/filter.tsv |
perl -nla -F"\t" -e 'print for @F' |
spanr cover stdin -o tests/S288c/cover.json
spanr stat tests/S288c/chr.sizes tests/S288c/cover.json -o stdout
Atha
gzip -dcf tests/Atha/links.lastz.tsv.gz tests/Atha/links.blast.tsv.gz |
linkr sort stdin -o tests/Atha/sort.tsv
linkr clean tests/Atha/sort.tsv -o tests/Atha/sort.clean.tsv
rgr merge tests/Atha/sort.clean.tsv -c 0.95 -o tests/Atha/merge.tsv
linkr clean tests/Atha/sort.clean.tsv -r tests/Atha/merge.tsv --bundle 500 -o tests/Atha/clean.tsv
linkr connect tests/Atha/clean.tsv -o tests/Atha/connect.tsv
linkr filter tests/Atha/connect.tsv -r 0.8 -o tests/Atha/filter.tsv
wc -l tests/Atha/*.tsv
# 4500 tests/Atha/clean.tsv
# 3832 tests/Atha/connect.tsv
# 3832 tests/Atha/filter.tsv
# 785 tests/Atha/merge.tsv
# 5416 tests/Atha/sort.clean.tsv
# 7754 tests/Atha/sort.tsv
cat tests/Atha/filter.tsv |
perl -nla -F"\t" -e 'print for @F' |
spanr cover stdin -o tests/Atha/cover.json
spanr stat tests/Atha/chr.sizes tests/Atha/cover.json -o stdout
fasr
fasr maf2fas tests/fasr/example.maf
fasr axt2fas tests/fasr/RM11_1a.chr.sizes tests/fasr/example.axt --qname RM11_1a
cargo run --bin fasr filter tests/fasr/example.fas --ge 10
fasr name tests/fasr/example.fas --count
fasr cover tests/fasr/example.fas
fasr cover tests/fasr/example.fas --name S288c --trim 10
fasr concat tests/fasr/name.lst tests/fasr/example.fas
fasr subset tests/fasr/name.lst tests/fasr/example.fas
cargo run --bin fasr subset tests/fasr/name.lst tests/fasr/refine.fas --required
fasr link tests/fasr/example.fas --pair
fasr link tests/fasr/example.fas --best
cargo run --bin fasr replace tests/fasr/replace.tsv tests/fasr/example.fas
cargo run --bin fasr replace tests/fasr/replace.fail.tsv tests/fasr/example.fas
samtools faidx tests/fasr/NC_000932.fa NC_000932:1-10
fasr check tests/fasr/NC_000932.fa tests/fasr/A_tha.pair.fas
fasr create tests/fasr/genome.fa tests/fasr/I.connect.tsv --name S288c
# Create a fasta file containing multiple genomes
cat tests/fasr/genome.fa | sed 's/^>/>S288c./' > tests/fasr/genomes.fa
samtools faidx tests/fasr/genomes.fa S288c.I:1-100
cargo run --bin fasr create tests/fasr/genomes.fa tests/fasr/I.name.tsv --multi
fasr separate tests/fasr/example.fas -o . --suffix .tmp
spoa tests/fasr/refine.fasta -r 1
cargo run --bin fasr consensus tests/fasr/example.fas
cargo run --bin fasr consensus tests/fasr/refine.fas
cargo run --bin fasr consensus tests/fasr/refine.fas --outgroup -p 2
cargo run --bin fasr refine tests/fasr/example.fas
cargo run --bin fasr refine tests/fasr/example.fas --msa none --chop 10
cargo run --bin fasr refine tests/fasr/refine2.fas --msa clustalw --outgroup
cargo run --bin fasr refine tests/fasr/example.fas --quick
cargo run --bin fasr split tests/fasr/example.fas --simple
cargo run --bin fasr split tests/fasr/example.fas -o . --chr --suffix .tmp
cargo run --bin fasr slice tests/fasr/slice.json tests/fasr/slice.fas --name S288c
cargo run --bin fasr join tests/fasr/S288cvsYJM789.slice.fas --name YJM789
cargo run --bin fasr join \
tests/fasr/S288cvsRM11_1a.slice.fas \
tests/fasr/S288cvsYJM789.slice.fas \
tests/fasr/S288cvsSpar.slice.fas
cargo run --bin fasr stat tests/fasr/example.fas --outgroup
cargo run --bin fasr variation tests/fasr/example.fas
cargo run --bin fasr variation tests/fasr/example.fas --outgroup
cargo run --bin fasr xlsx tests/fasr/example.fas
cargo run --bin fasr xlsx tests/fasr/example.fas --outgroup
cargo run --bin fasr pl-p2m tests/fasr/S288cvsRM11_1a.slice.fas tests/fasr/S288cvsSpar.slice.fas
许可证

依赖项
~20–32MB
~443K SLoC