17个不稳定版本 (5个重大变更)
| 0.6.0 | 2023年2月28日 |
|---|---|
| 0.5.0 | 2022年12月13日 |
| 0.4.3 | 2022年9月9日 |
| 0.4.1 | 2021年1月22日 |
| 0.1.0 | 2017年3月28日 |
#15 in 生物学
每月下载量776
在6个组件(5个直接)中使用
210KB
4.5K SLoC
Finch
![]()
![]()
Finch是基因组数据的Min-wise独立排列局部敏感哈希(MinHashing)实现。此仓库提供库和命令行接口,重新实现了One Codex的现有内部聚类/序列搜索工具(并添加了新功能/扩展!)使用Rust。
入门指南
安装
您可以从源代码构建Finch,这需要Rust >= 1.49。Rust的Cargo包管理器(有关Cargo安装说明,请参阅rustup)可以自动构建和安装Finch,使用cargo install finch_cli。如果您需要Python绑定,您必须采取额外步骤(请参阅Python支持)。或者,下载预构建的二进制文件或从PyPi pip install finch-sketch安装。
示例用法
要开始使用,我们首先为几个FASTA或FASTQ文件计算sketches。这些sketches是底层基因组数据的紧凑、采样表示,允许finch快速估计数据集之间的距离。使用finch sketch命令绘制文件
finch sketch example.fastq example2.fastq
生成的草图文件(example.fastq.sk 和 example2.fastq.sk)可以与其它 finch 命令一起使用(以及与其它 MinHash 实现1)。请注意,Finch 的所有命令都可以接受草图或原始序列文件。如果传递后者,finch 将在运行时绘制草图。然而,在运行时生成的草图不会被finch sketch保存,因此如果您打算多次使用草图,应该调用finch sketch。
草图完成后,可以比较多个测序运行以确定它们有多相似
finch dist example.fastq.sk example2.fastq.sk
这将打印出包含一些关键距离统计结果的结果(以 JSON 格式),包括 containment 和 jaccard 相似度得分以及一个 mashDistance 距离估计
[
{
"commonHashes": 30,
"containment": 0.03,
"jaccard": 0.015228426395939087,
"mashDistance": 0.1669789474914277,
"query": "example2.fastq",
"reference": "example1.fastq",
"totalHashes": 1000
}
]
在这种情况下,这些文件估计的距离约为 0.17,包含量为 0.03(即,这两个 FASTQ 共享 3% 的 min-mer)。请注意,使用更大的 --n-hashes 参数重新计算草图可以提供高度相似数据集的额外分辨率。
接下来,我们可能想要在 RefSeq 的所有基因组中找到与我们的示例 FASTQ 最接近的基因组。为此,我们只需将示例查询文件作为第一个参数传递,并将包含 RefSeq 中所有基因组的草图文件作为第二个参数传递(有关与 finch 一起使用的预计算 RefSeq 数据库的示例数据部分)
finch dist ./example.fastq.sk ./refseq_sketches_21_1000.sk --max-dist 0.2
在这里,我们还设置了最大距离为 0.2,以便过滤掉不太相关的基因组(距离为 0 表示完全相同的基因组)。设置最大距离可以确保只返回相关的结果--省略此参数将返回到 RefSeq 中所有基因组的距离。
注意:以下每个命令都将在下面详细介绍,并且还可以通过传递每个命令的 --help 标志来获取更多信息,例如,finch dist --help。
设计目标
我们对 Finch 有 3 个主要的设计目标
-
支持跟踪 k-mer/minmer 计数;
-
改进用于原始序列数据(即 FASTQ)的错误过滤;以及
-
改进性能。
支持计数
Finch 是第一个支持跟踪每个唯一 k-mer/minmer 丰度的 MinHash 实现。这除了支持更高级的过滤(在这里实现)和计数感知距离度量(一个未来研究区域)外,还允许对数据进行有用的快速解释(例如,使用 finch hist 可视化测序深度)。由于 Finch 还支持新的 Mash JSON 兼容格式,因此我们可以将此计数信息保存下来以供下游处理和比较。
⚠️ 注意,尽管 Finch 支持常见的 JSON 交换格式,但由于哈希种子值不兼容,可能仍然存在不兼容性(Finch 使用种子值
0,而 Mash 使用42;有关更多信息,请参阅 此问题)。
过滤
Finch的设计目标之一是能够准确且稳定地绘制细菌分离株的可变深度测序图(即,无需高质量组装)。虽然Mash包括指定固定绝对截止值(或使用Bloom过滤器过滤样本中的独特minmers)的功能,但实际上这种过滤机制在多样化的测序深度、基因组大小和测序错误模式上表现不佳。此处实现的Finch过滤方法扩展了原始Mash论文中的几个想法,以及关于聚类临床C. diff分离株的先前工作。
Finch包含两种自动过滤类别(默认开启对FASTQs的过滤)以应对这一挑战
-
基于计数的自适应过滤:我们的默认实现“过度草图”一个输入文件(即,包括比用户指定的多n倍的minmers),并动态确定一个合适的基于计数的过滤截止值,以去除低丰度的k-mers(这些主要是由随机错误组成)。作为备用,此实现从不排除超过可合理假设为错误的上限比例的minmers(
k倍指定的--err-filter错误率,默认为21%)。 -
定向过滤:我们还应用了一个“定向过滤器”,以去除在单一方向上大量存在的k-mers;实际上,这些通常是测序适配器或其他不反映底层样本的杂质(并可能错误地降低两个草图之间的相似度)。
实际上,这些优化显著提高了可变深度分离株与完成组装之间的距离估计的稳定性和准确性。下面的图表显示了高质量细菌组装与代表不同测序深度的FASTQ文件草图之间的距离:无过滤(cutoff=0),“天真”过滤(cutoff=1),以及Finch的自适应、可变截止过滤。我们使用包含度衡量标准,结果越接近100%越好
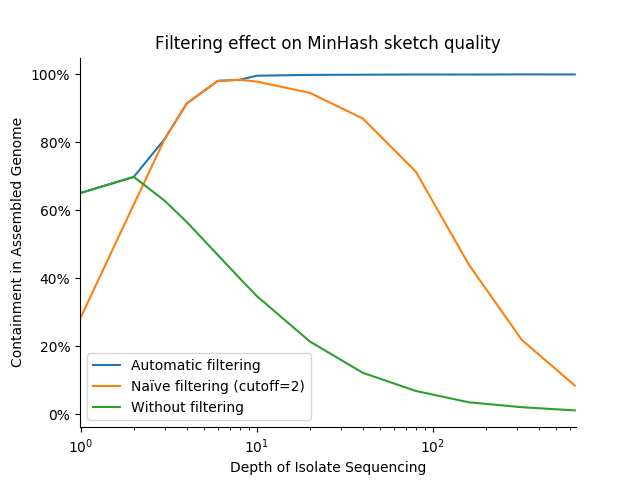
值得注意的是,这种策略在测序深度方面表现良好,而固定的截止值0和1从未达到近100%的预期包含度(而且前者在适度的深度上会失败得非常严重)。它对重复错误也很鲁棒(即,错误k-mers/minmers的计数>=2),这在测序深度低至10-20X时可能会开始成为一个问题。
速度/性能
除了Rust提供的某些内存安全性保证外,我们还在现有实现上看到了相当的速度提升。理想情况下,哈希应该是在MinHashing中的速率限制步骤。分析表明finch大约花费了三分之一的运行时间在murmurhash3_x64_128上,因此我们应该接近这个理论极限。
| Mash | Mash(过滤后) | Sourmash | Finch | Finch(过滤后) | |
|---|---|---|---|---|---|
| 时间 | 238秒 | 276秒 | 518秒 | 99秒 | 104秒 |
| 最大内存(MB) | 1.2 | 501.1 | 13.9 | 21.8 | 60.0 |
注意:基准测试在2015年初的MacBook Pro上进行。基准测试是草图化一个4.8GB的FASTQ文件(SRR5132341.fastq),草图大小为n=10,000。Finch过滤结果使用默认选项,而Mash使用
-b 500Mb来分配500MB的Bloom过滤器。基准测试使用了以下提交的Mash(23776db)和sourmash(5da5ee7)
用法
Finch支持四种主要操作,其中许多操作具有类似的参数。
共享参数
Finch可以接受多个参数来控制绘图的方式(这些选项也适用于dist、hist和info命令)。
-n <N>/--n-hashes <N>控制整个sketch的大小(更高的值在比较时提供更好的分辨率)。默认1000。-k <K>/--kmer-length <K>设置要散列的kmer的大小(更高的值使比较在分类学上更具体)。默认21。--seed <S>设置散列的种子。这只有在直接导出sketch与其他版本的Mash算法比较时才需要更改,这些算法使用非零默认种子。默认0。
绘图完成后,会进行过滤,可以通过多个选项进行控制
-f/--filter/--no-filter确定是否应用过滤(如果没有指定,默认对FASTQ文件进行过滤,不对FASTA文件进行过滤)--min-abun-filter <MIN>/--max-abun-filter <MAX>设置所有kmer必须存在的绝对最小和最大丰度(包含)。最小过滤器将覆盖任何自适应错误过滤器猜测(下面)。--err-filter <ERR_VALUE>是默认的自适应过滤方案。从概念上讲,ERR_VALUE应该是测序仪的错误率或更高。请注意,高错误率测序数据(例如长读测序)通常在MinHashing中表现不佳。--strand-filter <V>设置链过滤器截止值。
请注意,如果没有足够的kmer来自oversketch以满足sketch大小,绘图将失败。有两个选项可能有所帮助
--oversketch <N>可以用来增加oversketch的大小(通常是200倍),并增加过滤后的版本足够大的可能性。--no-strict将允许Finch使用小于指定大小的sketch进行操作。请谨慎使用。
finch sketch
finch sketch将读取FASTA或FASTQ文件并生成一个“sketch”,可用于进一步的操作。
如果正在读取的文件是sketch并且已设置过滤标志(-f/--filter),则将对sketch重新应用丰度过滤器,以便进行post-sketch过滤。链过滤器只能应用于原始FAST(A/Q)文件,因为当保存sketch时,链级别数据会丢失。
默认情况下,sketch会在绘制的FAST(A/Q)文件旁边生成带有.sk后缀的文件。可以通过传递-O(大写O)将其写入标准输出,或者传递-o <文件>(小写o)将其写入文件。要从标准输入读取,请使用文件名-;这允许将文件流式传输到finch,例如cat testfile.fq | finch sketch -o testfile.sk -。
如果编辑它们的src/mash/hash.h并将哈希值设置为0,或者通过设置--seed 42手动覆盖Finch的种子值,则绘制的草图应该与原始Mash实现兼容。
finch dist
finch dist将计算不同草图之间的Jaccard距离。如果列出的文件是FASTA/Q而不是草图,Finch将自动使用与sketch相同的命令行参数将它们绘制到内存中,或者对于第一个文件之后的文件,使用绘制第一个文件时使用的参数。
距离和包含度将仅从查询集合中的一个或多个查询计算;默认情况下,列表中的第一个草图将用作查询,所有其他草图将用作参考。可以通过传递--queries <sketch_1>,<sketch_2手动覆盖此行为,并可以使用其他草图作为参考。此外,传递--pairwise选项将计算所有草图之间的距离。
由于不同的计数算法和停止标准,距离可能与原始Mash程序和较旧版本的Finch中的计算略有不同。通过传递-old-dist标志将回退到Finch的较旧版本的计算;自版本0.3起已取消对Mash精确距离计算的支持。
finch hist
finch hist将为每个提供的草图输出JSON格式的直方图。直方图是每个深度上minmers数量的列表,例如,对于有两个minmers的草图,一个深度为1(第一个位置),一个深度为3(第三个位置),表示为{"sketch_name": [1, 0, 1]}。
⚠️ 您可以使用以下命令与Matplotlib一起快速生成直方图:
finch hist test.fastq.sk | python -c 'import json; import matplotlib.pyplot as plt; import sys; v = json.loads(sys.stdin.read()).values()[0]; plt.plot(range(1, len(v)+1), v); plt.show()'。请注意,finch hist的JSON输出可能在未来的版本中改变,例如,改为更紧凑的{count: value}格式或类似格式。
finch info
finch info将为每个提供的sketch输出一个格式化的列表,包括% GC,覆盖率等。
⚠️ 请注意,此返回的值是近似的,并且用于计算的算法仍然是粗糙的,可能随时更改。
示例数据
我们已经使用此脚本绘制了NCBI RefSeq集合(截至2017年3月27日),并为每个细菌和病毒基因组创建了带有单独sketch的tar包。链接:k=21和n=1,000,k=31和n=1,000,k=21和n=10,000,以及k=31和n=10,000。
参考文献 & 注释
还有其他几种Mash算法的实现,应与本实现兼容/可比较,特别是
注释
- 1 但是,请参阅此问题跟踪互操作性,并请注意,其他实现可能使用不同的种子值。
Python支持
您可以使用Pip安装Finch
pip install finch-sketch
您可以使用以下命令将Finch编译为Python库
pip install maturin
cd lib
maturin build --features=python --release --strip
# or maturin develop, etc
# to cross-compile linux wheels:
cp ../README.md .
# and edit the `readme` key of Cargo.toml
docker run --rm -v $(pwd):/io ghcr.io/pyo3/maturin:main build --features=python --release --strip --interpreter=python3.10 --compatibility=manylinux2010
然后,例如,要计算两个大肠杆菌之间的相似性
from finch import sketch_file
sketch_one = sketch_file('WIS_EcoB_v2.fas')
sketch_two = sketch_file('WIS_Eco10798_DRAFTv1.fas')
cont, jacc = sketch_one.compare(sketch_two)
Cap'n Proto
在src/serialization文件中有finch.capnp,以及MinHash模式输出的内容(https://github.com/marbl/Mash/blob/54e6d66b7720035a2605a02892cad027ef3231ef/src/mash/capnp/MinHash.capnp)
- 由 @bovee 在 https://github.com/bovee/Mash/blob/master/src/mash/capnp/MinHash.capnp 中做出的更改
这两个都是在安装了 capnp 和通过以下命令使用 cargo install capnpc 后生成的
capnp compile -orust:finch-lib/src/serialization/ --src-prefix=finch-lib/src/serialization/ finch-lib/src/serialization/finch.capnp
capnp compile -orust:finch-lib/src/serialization/ --src-prefix=finch-lib/src/serialization/ finch-lib/src/serialization/mash.capnp
然后搜索并替换 crate:: 路径以修复它 crate::serialization::。
贡献
可以通过 GitHub 的问题报告来报告问题或改进建议。我们很高兴接受和/或指导任何新增或修复(最好以拉取请求的形式提交)。关于我们的行为准则,请参阅 CODE_OF_CONDUCT.md。
依赖项
~4–11MB
~118K SLoC