7个版本
| 0.3.4 | 2022年10月29日 |
|---|---|
| 0.3.3 | 2022年10月28日 |
| 0.2.0 | 2022年8月18日 |
| 0.1.0 | 2022年8月18日 |
#135 in 生物学
每月下载量 34
2MB
880 行
二倍体污染
![]()
![]()
一个模块,用于从二倍体变异调用中估算污染水平。这是大量受到 Dcon 的启发。
背景
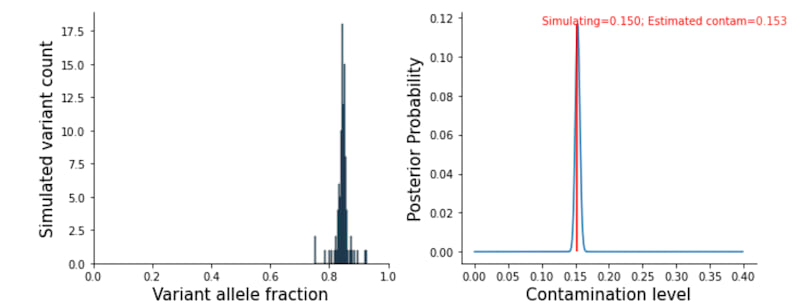
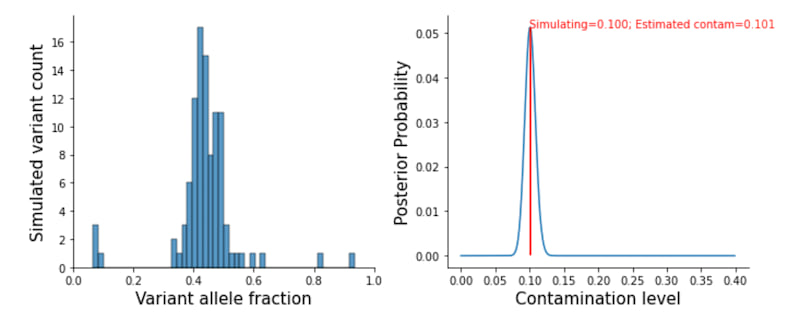
我们假设污染水平将在 0-0.4 之间,并且在每个假设的污染水平下,我们将使用二项模型计算观察到观察到的变异等位基因频率的概率,假设给定的污染水平对于 VCF 文件中的每个变异都是真实的。然后我们计算所有变异的对数似然并选择最大似然污染水平作为最终调用。
伪代码
n = total_count
x = alt_allele_count
contam = 0
max_log_likelihood = -inf
for contam_level in all_contamination_level:
p = expected_alt_fraction_for_the_given_contamination_level
log_likelihood = sum(binom_loglik(n, x, p) for all_variants)
if log_likelihood > max_log_likelihood:
contam = contam_level
在此 处 进行的一个模拟研究。
同型变异
对于同型变异,在给定的污染水平 $c \in [0,0.4]$ 下,观察到的期望变异等位基因计数 $x$ 的概率为 $n$ 的读深度
$$ P(X=x,c) = \binom{n}{x}p^x(1 - p)^{n-x} $$
其中 $p = (1-c)$ 在所有同型变异中
杂合变异
对于杂合变异,在给定的污染水平 $c \in [0,0.4]$ 下,观察到的期望变异等位基因计数 $x$ 的概率将遵循上述二项分布,但 $p$ 可以是
- 当由于污染观察到低等位基因频率时,$(1 - c)/2$
- 当由于污染将同型变异调用为杂合变异时,$(1 - c)$
- 当污染看起来像等位基因时,$(0.5 + c)$,使得等位基因频率高于预期
- 当污染看起来像参考等位基因时,$(0.5 - c)$,使得等位基因频率低于预期
- 当污染本身被调用为低变异频率杂合变异时,$c$
在评估这些情况后,我们将从给定污染水平对数似然的总和中选择最高概率的事件。
Rust
我们还用 Rust 编写了代码。
要运行代码
$ cargo run -- -i data/test.vcf -d debug_json
或
$ cargo install --path .
$ target/release/diploid-contam-estimator
Douglas Wu <wckdouglas@gmail.com>
Estimating contamination level from a diploid VCF file
The program assume we are dealing with a diploid genome, and using the
deviation of allelic balance from the expected allelic frequence for homozygous
or heterozygous variant calls to compute a contamination value.
For homozygous variants, we deviation from allelic frequency of 1 is all introduced by
contaminaion.
For heterozygous variants, it is a little more complex, because it could be due to:
1. contamination that doesn't look like the HET ALT allele: we expect lower HET alt allele
frequency
2. contamination that doesn't look like the HOM ALT allele: we expect High HET alt allele
frequency
3. contamination that looks like the ALT allele: we expect higher alt allele frequency
4. contamination that looks like the REF allele: we expect lower alt allele frequency
5. contamination being called as ALT
USAGE:
diploid-contam-estimator [OPTIONS] --in-vcf <in_vcf>
OPTIONS:
-d, --debug-json <debug_json>
A json output file for storing all intermediate log prob
-h, --help
Print help information
-i, --in-vcf <in_vcf>
A diploid vcf file for estimating contamination
-m, --min-depth <depth_threshold>
Minimum depth for a variant to be considered (i.e. DP tag) [default: 0]
-o, --out-json <out_json>
A json output file for storing the maximum likelihood contam level for the vcf file
--snv-only
Only use SNV (ignore indel) for contamination estimations
-v, --debug-variant-json <debug_variant_json>
A json output file for storing all input variants used for calculation
-V, --version
Print version information
依赖关系
~10–20MB
~268K SLoC