11 个版本 (7 个稳定版本)
| 1.2.1 | 2024年4月21日 |
|---|---|
| 1.1.0 | 2023年2月9日 |
| 1.0.3 | 2022年7月22日 |
| 1.0.1 | 2022年3月5日 |
| 0.2.1 | 2021年6月27日 |
#136 在 算法
520KB
12K SLoC
biodiff
![]()
使用比对算法比较二进制文件。
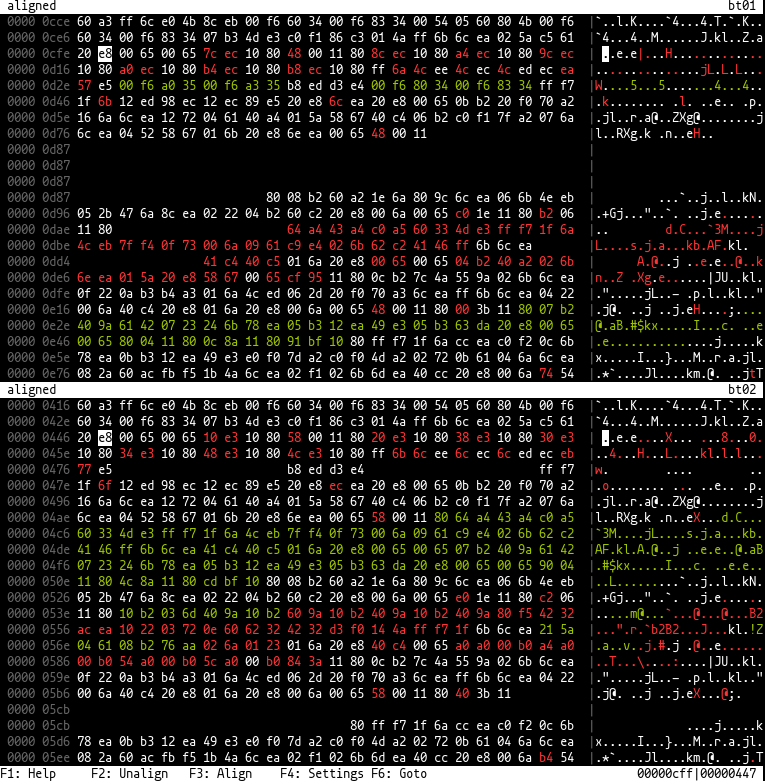
这是什么
这是一个二进制差异工具。
该工具可以将两个二进制文件并排显示,以便相似位置在两侧处于相同位置,并且从一侧缺失的字节将用空字节填充。它使用来自 'wfa2' 或 rust-bio 库(通常用于 DNA 序列比对)的生物信息学算法。配置对话框使用 cursive 实现。
特性
- 非对齐视图,允许独立移动两侧作为连续的字节段
- 对齐视图,用于比较两个文件的对应字节
- 许多可配置的字节表示(基数 2、8、10、16;混合 ASCII/十六进制、盲文、罗马数字)
- 从右到左模式,水平和垂直分割,ASCII 和条形列
- 每行字节数可配置,可以通过按
[,],0调整 - 通过按 '=' 查找可见/选择的字节中的重复项来自动确定宽度
- 使用文本、正则表达式和十六进制搜索
用法
在终端中执行 biodiff file_a file_b,你应该会进入一个显示两个文件并排的十六进制视图。最初,文件将不会对齐,并在每侧无间隔地显示。通过将光标和视图移动到左右两侧相似的位置,并按 F3(或 3),它们可以对齐。这是在标准配置下按块进行的,这意味着靠近光标的字节首先对齐,稍后显示在两侧进一步对齐的块。
您还可以通过使用 F4(请务必查阅参数的帮助信息)来更改设置,从而进行全局对齐(一次对齐整个文件)。由于它需要二次时间和空间复杂度,因此全局对齐可能不适合大于64KB的文件。还有一种“带状”算法,速度更快,但精度略低。
您还可以选择一个文件上的区域,并通过按F3,对齐算法将使用所选字节作为模式来在另一个文件上找到相应的字节,进行半全局对齐。
您还可以通过使用 biodiff --print file_a file_b 将diff直接打印到终端。在这种情况下(如果文件足够小,不会花费太多时间),您可以添加 -gglobal 标志来进行全局对齐(与适合交互式使用的块状对齐相反)。
安装
如果您幸运的话,您的默认包管理器可能会有一个包可用,请参阅repology页面。在发布页面下,应该有适用于某些环境的可下载的二进制文件。或者,您也可以通过执行 cargo install biodiff 来使用 cargo 安装。您需要安装 cmake 才能编译 wfa2 功能。请注意,如果您使用 Windows,并且想要 wfa2 支持,则需要使用 x86_64-unknown-linux-gnu 目标。
您还可以通过执行此存储库中的代码直接执行,通过执行 cargo run --release -- file_a file_b。请注意,配置文件仅在标记发布之间保证兼容性。
默认情况下,设置存储在平台特定的用户目录中。要使用自定义设置目录,请在运行 biodiff 之前将环境变量 BIODIFF_CONFIG_DIR 设置为所需的目录路径。如果目录不存在,它将被自动创建。
许可证
本项目采用MIT许可证。
依赖项
~24–37MB
~518K SLoC