11个版本
| 0.2.0 | 2022年9月22日 |
|---|---|
| 0.1.9 | 2022年9月13日 |
#3 in #seq
225KB
211 行
secondary_rewriter
一些对齐器,如minimap2,不会将SEQ和QUAL字段写入次级对齐(有时称为多映射器,参见SAMv1.pdf中的多重映射 https://samtools.github.io/hts-specs/SAMv1.pdf),这使得分析它们变得困难(例如,次级对齐中的SNPs在基因组浏览器中不可见,并且变异调用不会在这些对齐上工作)。此程序向次级对齐添加SEQ/QUAL,通过参考主对齐来获取SEQ和QUAL。
Minimap2参考 https://github.com/lh3/minimap2/issues/458 https://github.com/lh3/minimap2/pull/687
安装
首先安装rust,可能使用rustup https://rustup.rs/
然后
cargo install secondary_rewriter
用法
此小型shell脚本自动执行多步骤管道(支持BAM或CRAM)
#!/bin/bash
# write_secondaries.sh
# usage
# ./write_secondaries.sh <input.cram> <ref.fa> <output.cram> <nthreads default 4> <memory gigabytes, per-thread, default 1G>
# e.g.
# ./write_secondaries.sh input.cram ref.fa output.cram 16 2G
THR=${4:-4}
MEM=${5:-1G}
samtools view -@$THR -h $1 -T $2 -f256 > sec.txt
samtools view -@$THR -h $1 -T $2 -F256 | secondary_rewriter --generate-primary-loc-tag --secondaries sec.txt | samtools sort --reference $2 -@$THR -m $MEM - -o $3
两遍策略
两遍策略的工作原理如下
- 第一遍:将所有次级对齐(具有标志256的读取)输出到外部文件
- 第二遍:从外部文件读取次级对齐到内存中,然后扫描原始SAM/BAM/CRAM,将主对齐的SEQ和QUAL字段添加到次级对齐中,并管道到
samtools sort(由于所有次级读取都将顺序混乱,添加到找到SEQ/QUAL的主对齐之后,这是必需的)
此过程避免了将整个SAM/BAM/CRAM加载到内存中,但确实需要samtools sort,这是一个有点昂贵的操作。尽管如此,它确实将所有次级对齐(在添加SEQ/QUAL字段之前,通常为几个吉字节而不是数百吉字节)加载到内存中。
结果
您的次级读取现在将显示SNPs等,并在基因组浏览器中显示。具有SEQ对于变异调用也很重要。
IGV和JBrowse 2的截图(仅为了表明它不是浏览器特定的),显示了在基因组的一个区域(着丝粒区域的超长读hs37d5 https://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/data/AshkenazimTrio/HG002_NA24385_son/Ultralong_OxfordNanopore/guppy-V2.3.4_2019-06-26/)中具有许多次级对齐的基因组区域的文件在运行secondary_rewriter之前和之后的相同文件。
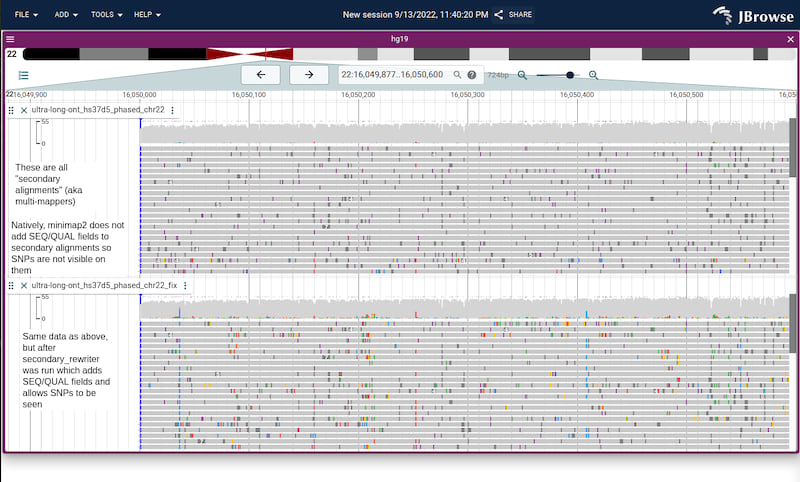 在JBrowse 2中运行secondary_rewriter之前/之后的截图
在JBrowse 2中运行secondary_rewriter之前/之后的截图
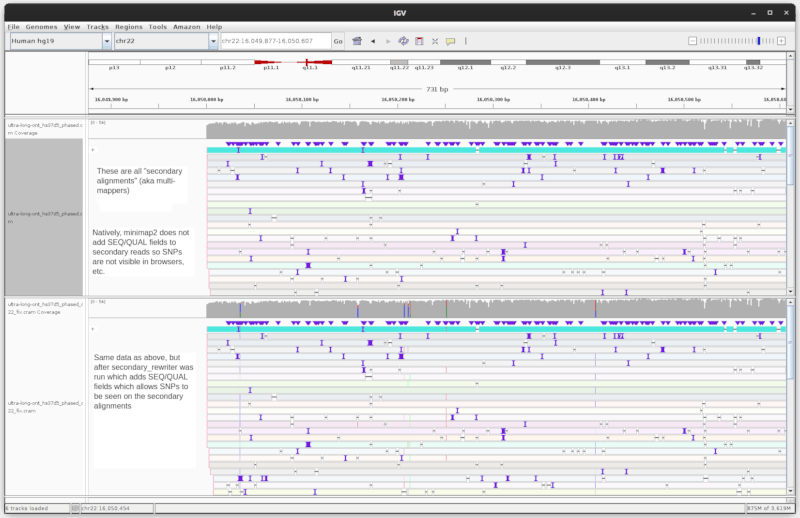 IGV中的相同数据截图,前后对比
IGV中的相同数据截图,前后对比
帮助
secondary_rewriter 0.1.9
Adds SEQ and QUAL fields to secondary alignments from the primary alignment
USAGE:
secondary_rewriter [OPTIONS]
OPTIONS:
-g, --generate-primary-loc-tag Boolean flag on whether to produce a tag like pl:Z:chr1:1000
on the secondary alignments that says where the primary
alignment is
-h, --help Print help information
-s, --secondaries <SECONDARIES> Path to file of secondary reads (generated by e.g. samtools
view -f256)
-V, --version Print version information
--generate-primary-loc-tag 在二级读段上创建标签,例如 pl:Z:chr1:1000,以指出主读段的位置
运行时间
该程序的速度主要受samtools view/samtools sort效率的限制,因此如果您为samtools提供大量的线程和内存,您的性能将得到提高。
在一个t2.2xlarge AWS实例上,使用8个线程和每线程1GB排序内存,对218GB的BAM文件(来自https://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/data/AshkenazimTrio/HG002_NA24385_son/Ultralong_OxfordNanopore/guppy-V2.3.4_2019-06-26/的ultra long read hs37d5)运行secondary_rewriter需要189分钟(约3小时)
注意,输出数据文件也更大,本例中结果为267Gb BAM,而原始为218Gb BAM。
可能考虑事项
-
可能存在minimap2不输出这些字段的原因(作者引用输出大小),但完全可以添加回SEQ和QUAL。此PR到minimap2原生输出SEQ和QUAL https://github.com/lh3/minimap2/pull/687/files,但已经声明minimap2 "不会" 输出这些。
-
此程序不处理CIGAR中的硬剪辑(
H操作符),但我还没有看到minimap2的输出。如果您看到这个问题,请让我知道,可能可以修复。 -
最后,您可能还需要考虑如何在您的流程中处理二级比对的影响,尽管这个程序可以帮助在特定情况下,但这些二级/多比对对的影响可能不清楚。
依赖
~3.5MB
~66K SLoC