8 个版本
| 0.3.2 | 2022年9月16日 |
|---|---|
| 0.3.1 | 2022年9月7日 |
| 0.2.6 | 2021年10月3日 |
| 0.2.3 | 2021年2月17日 |
| 0.1.1 | 2020年8月7日 |
#146 在 科学
9.5MB
2K SLoC
GeNovo - 识别具有新突变疾病的基因
每个人的基因组中都包含一些新突变:这些突变不是其父母基因组的一部分。这些新突变可能导致疾病,但仍然很难将致病的突变与那些与疾病无关的突变区分开来。此外,某些基因有更高的突变几率,这意味着即使在多个患病者中发生突变,该基因的突变也不足为奇。
GeNovo这个名称是“基因”检测基于“新发现”突变的双关语。
安装
首先确保你已经安装了 cargo(cargo 是Rust编程语言的包管理器)。最简单的方法是安装 rustup,它会自动为你安装 cargo 和 rustc。如果你使用的是基于UNIX的系统,你可能也能通过系统的包管理器安装 cargo 或 rustup。
其次,你需要运行
cargo install genovo
这将安装程序genovo到特定位置,该位置将在安装过程结束时显示。(你可能需要调整你的 PATH 变量,以便在命令行上自动找到程序)
该管道是一个单独的可执行文件(genovo),它结合了我之前组合的早期管道中的重要步骤。(单个可执行文件比所有松散耦合的脚本更容易使用。)
管道
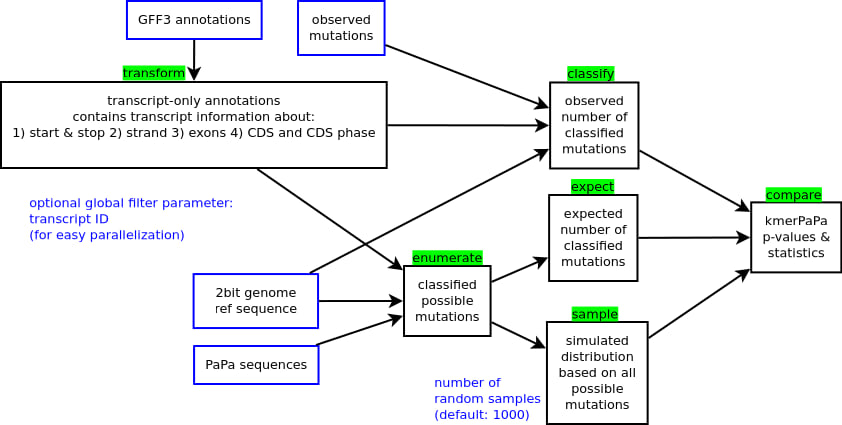
- 输入文件用蓝色边框标记。
- 管道步骤用黑色边框标记,其名称用绿色背景标记
- 可选参数用蓝色字体标记
输入和输出文件
该管道接受多个参数,代表输入和输出参数。根据正在执行的操作,参数可能被视为输入或输出文件。
例如,当指定--action expect时,--possible-mutations参数被视为输入文件。但是,当指定--action enumerate时,它被视为输出文件。如果没有指定--action参数,则整个管道运行,所有文件参数都视为输出文件。
您还可以将-指定为输入/输出文件。根据操作,这将解释为STDIN或STDOUT。
如果您担心空间使用,还可以指定.gz文件扩展名。genovo将自动解压缩/压缩输入/输出。
ID过滤/并行处理
如果传递了参数--id ID,则仅处理具有ID ID的转录本,其他所有转录本将被忽略。这对于并行执行很有用。您可以针对每个转录本启动一个单独的任务。请注意,--id仅接受单个ID。不支持多个ID。您可以将--id选项与其他任何选项结合使用。
在整个测试数据集上运行整个管道
您可以从git仓库下载测试数据集
wget https://github.com/BesenbacherLab/genovo/raw/master/test_data/test_data.tar.gz
tar xvzf test_data.tar.gz
如果您想在测试数据上运行整个管道,可以执行以下命令
genovo \
--gff3 gencode.v19.CCDS.annotation.protein_coding_chr22.gff3.gz \
--observed-mutations observed_mutations_DDD_2017_chr22.txt \
--genome hg19_chr22.2bit \
--point-mutation-probabilities SNV_mutation_rate_model.txt \
--indel-mutation-probabilities indel_mutation_rate_model.txt \
--scaling-factor 8586 \
--significant-mutations genovo_results_chr22.txt
该命令在“正常”计算机上运行需要约1分钟。它将观察到的、预期的和采样的突变之间的比较写入文件genovo_results_chr22.txt。如果没有使用--significant-mutations参数,结果将写入STDOUT。对于每个转录本t和突变类型m,输出包含以下输出列
| 列 | 描述 |
|---|---|
| 观察到的 | 转录本t中观察到的m突变数 |
| 预期的 | 根据突变率模型,转录本t中预期的m突变数 |
| 预期下限 | 围绕预期突变数的置信区间下限 |
| 预期上限 | 围绕预期突变数的置信区间上限 |
| p值 | 针对基于抽样的测试的单侧p值,测试转录本t中是否存在比预期更多的观察到的类型为m的突变 |
在测试三联体的de novo突变时,应将--scaling-factor设置为数据中三联体数量的两倍。因此,在这种情况下,它设置为8586,因为具有观察到的突变的数据集在4293个儿童中寻找de novo突变。
SNV_mutation_rate_model.txt和indel_mutation_rate_model.txt是使用kmerpapa创建的突变率模型。可以从https://github.com/BesenbacherLab/Genovo_Input下载正确输入格式的训练模型。
管道步骤
可以通过指定--action STEP参数独立执行管道的每个步骤。如果没有指定--action参数,则运行所有步骤(如上述示例所示)。
转换
此步骤接受一个gff3文件并将其转换为regions文件(这是一个特定于genovo的文件格式)。gff3文件需要以下属性
- ID:用于识别每个转录本的名字
- Parent:用于识别外显子和CDS区域的父元素
文件还必须排序,以确保所有外显子和CDS区域都列在其转录本行之后。
一些示例gff3文件可以从GENCODE获取。
强烈建议您只使用仅包含编码转录本的注释。非编码转录本的结果信息量较小。
参数
| 参数 | 输入/输出 | 描述 |
|---|---|---|
| --gff3 | 输入 | 包含转录本、外显子和CDS信息的GFF3注释文件 |
| --genomic-regions | 输出 | 将GFF3条目转换为变换版本,仅限于转录本、外显子和CDS区域 |
枚举
此命令遍历每个转录本的基因组序列,确定所有可能的单点突变及其概率。此命令需要一个PaPa-rates文件,该文件表示不同序列上下文中的单点突变概率,如我们在论文《使用k-mer模式分区改进特定位置突变率预测》中所述。此命令还需要一个twobit文件格式的参考基因组文件。请确保所有输入文件使用相同的基因组构建!
输出文件相当简单
#transcript_ID
numeric_mutation_type:mutation_probability
numeric_mutation_type:mutation_probability
numeric_mutation_type:mutation_probability
#other_transcript_ID
numeric_mutation_type:mutation_probability
numeric_mutation_type:mutation_probability
numeric_mutation_type:mutation_probability
numeric_mutation_type:mutation_probability
numeric_mutation_type:mutation_probability
numeric_mutation_type:mutation_probability
numeric_mutation_type:mutation_probability
每个转录本的可能突变以一个#ID行开始,然后是每个可能突变的行,其中概率不是NaN。请注意,每个位置都可能突变成另外三个碱基之一,因此每个位置都有三个可能的单点突变。
数值突变类型定义在突变类型数值代码表中。
参数
| 参数 | 输入/输出 | 描述 |
|---|---|---|
| --genomic-regions | 输入 | 将GFF3条目转换为变换版本,仅限于转录本、外显子和CDS区域 |
| --genome | 输入 | A twobit参考基因组序列文件 |
| --point-mutation-probabilities | 输入 | 包含序列上下文相关的点突变概率的模式分区文件 |
| --indel-mutation-probabilities | 输入 | 包含序列上下文相关的indel概率的模式分区文件 |
| --possible-mutations | 输出 | 每个转录本的可能突变 |
分类
此步骤对观察到的单点突变(这是您应提供的原始输入文件)进行分类。输入文件格式如下
chr1 1230448 G A
chr1 1609723 C T
chr1 1903276 C T
chr1 2574999 C T
第一列是染色体(必须与注释和twobit文件中的命名约定相同)。第二列是染色体上的位置。此位置必须是1基于的。第三列是参考基。如果此参考基与twobit文件中的内容不匹配,您将收到错误。第四列是新突变到的基。
参数
| 参数 | 输入/输出 | 描述 |
|---|---|---|
| --observed-mutations | 输入 | 您自己的数据中的观察到的点突变 |
| --genome | 输入 | A twobit参考基因组序列文件 |
| --genomic-regions | 输入 | 将GFF3条目转换为变换版本,仅限于转录本、外显子和CDS区域 |
| --classified-mutations | 输出 | 观察到的突变。但用数值突变类型进行了注释 |
预期
此步骤汇总所有可能突变及其概率,并给出表示每个基因预期突变数的总数。
参数
| 参数 | 输入/输出 | 描述 |
|---|---|---|
| --possible-mutations | 输入 | 每个转录本的可能突变 |
| --expected-mutations | 输出 | 每个转录本每种突变类型的预期突变数 |
样本
此步骤随机选择可能突变,并根据其概率随机模拟它们是否发生。这创建了一个用于在后续步骤中计算p值的经验分布的抽样突变分布。
参数
| 参数 | 输入/输出 | 描述 |
|---|---|---|
| --possible-mutations | 输入 | 每个转录本的可能突变 |
| --number-of-random-samples | 整数 | 有多少随机样本?默认=1000 |
| --sampled-mutations | 输出 | 抽样突变的分布 |
比较
比较观察到的、预期的和抽样突变,以确定在转录本中偶然观察到那么多突变的p值。请注意,输出文件包含所有统计测试的结果,即使是那些不显著的。请注意,p值没有考虑到预期突变数。
参数
| 参数 | 输入/输出 | 描述 |
|---|---|---|
| --classified-mutations | 输入 | 观察到的突变。但用数值突变类型进行了注释 |
| --expected-mutations | 输入 | 每个转录本每种突变类型的预期突变数 |
| --sampled-mutations | 输入 | 抽样突变的分布 |
| --significant-mutations | 输出 | 不同突变类型的观察到的、预期的、抽样和p值表。 |
突变类型数值代码表
流水线的输入和输出文件几乎全部是文本文件。为了更节省空间,对不同的突变类型进行了数字编码。
| 突变类型名称 | 数字代码 |
|---|---|
| 未知 | 0 |
| 同义突变 | 1 |
| 错义突变 | 2 |
| 无义突变 | 3 |
| 终止密码子丢失 | 4 |
| 起始密码子丢失 | 5 |
| 标准剪接位点丢失 | 6 |
| 内含子内的突变 | 7 |
| 非移码插入或缺失 | 8 |
| 移码插入或缺失 | 9 |
依赖关系
~5MB
~81K SLoC