3 个不稳定版本
| 0.1.3 | 2024年5月13日 |
|---|---|
| 0.1.2 | 2023年5月1日 |
| 0.1.1 |
|
| 0.1.0 |
|
| 0.0.12 | 2022年10月29日 |
#4 in 生物学
278 每月下载次数
3MB
4.5K SLoC
GSearch:基于各种 MinHash-like 指标和 HNSW 的超快速可扩展基因组搜索
![]()
GSearch 代表 基因组搜索。
这个包(目前处于开发中)计算细菌和古菌(或病毒和真菌)基因组的 MinHash-like 签名,并将细菌的 id 和 MinHash-like 签名存储在 Hnsw 结构中以搜索新的请求基因组。在这个仓库中提供了大约 50,000 到 60,000 行高度优化的 Rust 代码,以及为这个仓库开发的几个其他 crate/libraries,例如 kmerutils、probminhash、hnswlib-rs 和 annembed,详情见下文。其中一些库非常受欢迎,已被使用约 10,000 次,见 此处。
此包由 Jean-Pierre Both jpboth 负责软件部分,由 Jianshu Zhao 负责基因组部分开发。我们还在这个仓库上创建了一个镜像 GitLab 和 Gitee(您需要先登录才能查看内容),以防在某些地区无法使用 GitHub 服务。
关键功能
基因组/tohnsw 的草图,构建 hnsw 图数据库
目标是使用 Jaccard 索引作为突变率或平均核苷酸身份(ANI)或平均氨基酸身份(AAI)的准确代理,根据以下方程(泊松模型或二项模型):$$ANI=1+\frac{1}{k}log\frac{2*J}{1+J}$$
或
$$ANI=(\frac{2*J}{1+J})^{\frac{1}{k}}$$
其中 J 是类似于 Jaccard 的指数(例如 ProbMinHash 中的 Jp 或 SuperMinHash、SetSketch 或 Densified MinHash 中的 J,属于局部敏感哈希算法的一种,适用于最近邻搜索,详见下文)而 k 是 k-mer 的大小。为了实现这一点,我们使用了 sketching 技术。我们在基因组 DNA 或氨基酸序列中生成 k-mer,并在文件中绘制遇到的 k-mer 分布(可以是加权的或非加权的),详见 kmerutils。然后,最终的 sketch 存储在 Hnsw 数据库中,请参阅此处 hnsw 或此处 hnsw。
sketching 和 HNSW 图数据库构建是通过子命令 tohnsw 完成的。
Jaccard 指数有两种形式
1. 考虑 Kmer 重复性的概率 Jaccard 指数。它由以下公式定义:$$J_{P(A,B)}=\sum_{d\in D} \frac{1}{\sum_{d'\in D} \max (\frac{\omega_{A}(d')}{\omega_{A}(d)},\frac{\omega_{B}(d')}{\omega_{B}(d)})}$$ 其中 $\omega_{A}(d)$ 是 d 在 A 中的重复性(详见 Moulton-Jiang-arxiv)。在这种情况下,对于 J_p,我们使用在 probminhash 中实现的 probminhash 算法,或参阅原始论文/实现 此处。2. 无权(简单)的 Jaccard 指数由以下公式定义:$$Jaccard(A,B)=\frac{A \cap B}{A \cup B}$$ 在这种情况下,对于 J,我们使用 SuperMinHash、SetSketch(基于第 3.3 节中的局部敏感性或第 3.2 节中的联合最大似然估计,联合估计)或基于 One Permutation Hashing with Optimal Densification 方法的 densified MinHash 或基于 One Permutation Hashing with Faster Densification 方法,这些方法也已在上面提到的 probminhash 中实现。
上述提到的 sketching 选项可以通过 "gsearch tohnsw" 子命令中的 --algo 选项进行指定(prob、super/super2、hll 和 optdens)。我们建议使用 ProbMinHash 或 Optimal Densification。对于 SuperMinhash,有两个实现:super 是用于浮点类型 sketch 的,而 super2 是用于整数类型 sketch 的。后者比前者快得多。hll 是用于 SetSketch 的,我们将其命名为 hll,因为 SetSketch 数据结构可以被视为类似 HyperLogLog 的实现,但添加了新的相似性估计算法(例如,局部敏感性、LSH 或联合最大似然估计,JMLE 用于 Jaccard 指数),除了区分元素计数外。还因为我们认为 HyperLogLog 是一个美丽的名字,它描述了实现的关键步骤。
估计的类似于 Jaccard 的指数用于构建 HNSW 图数据库,该数据库由 crate hnswlib-rs 实现。
参考基因组 sketching 和 HNSW 图数据库构建可能需要一些时间(对于参数提供正确 sketching 质量的 GTDB 中约 65,000 个细菌基因组,少于 0.5 小时,或者对于整个 NCBI/RefSeq 原核生物基因组,约 318K,需要 2 到 3 小时)。结果存储在 2 个结构中
- 一个 Hnsw 结构,用于存储处理数据的排名和相应的 sketches。
- 一个字典,将每个排名与一个 fasta id 和 fasta 文件名关联。
Hnsw 结构被导出到 hnswdump.hnsw.graph 和 hnswdump.hnsw.data。字典被导出到一个 json 文件 seqdict.json。
将基因组添加到现有/预构建数据库中
对于将新基因组添加到现有数据库中,使用的是 add 子命令。它将自动加载 sketching 文件、图形文件以及构建图形时使用的参数,然后使用相同的参数将新基因组添加到现有数据库的基因组中。
请求、搜索新的基因组与预构建数据库
对于请求,使用子命令request。它重新加载已导出的文件,hnsw和seqdict相关操作需要一份包含请求的fasta文件列表,并为每个fasta文件根据上述距离导出所需数量的最近邻。一个包含3个关键列的表格文件将被保存到磁盘(gsearch.neighbors.txt)中:查询基因组路径、数据库基因组路径(按距离排序)和距离。距离可以按照上述公式转换为ANI或AAI。我们提供了程序reformat(也包括并行实现)来完成这个操作。
reformat -h
Processes input files for ANI calculation
Usage: reformat <kmer> <model> <input_file> <output_file>
Arguments:
<kmer> The kmer value used for ANI calculation (16)
<model> The model to be used for ANI calculation (1 or 2,corresponding to Poisson model and Binomial model)
<input_file> File containing the data to be transformed into tabular format
<output_file> File where the tabular output will be saved
Options:
-h, --help Print help
-V, --version Print version
reformat 16 1 ./gsearch.neighbors.txt ./clean.txt
然后您将看到如下整洁的输出:
| Query_Name | Distance | Neighbor_Fasta_name | Neighbor_Seq_Len | ANI |
|---|---|---|---|---|
| test03.fasta.gz | 5.40E-01 | GCF_024448335.1_genomic.fna.gz | 4379993 | 97.1126 |
| test03.fasta.gz | 8.22E-01 | GCF_000219605.1_genomic.fna.gz | 4547930 | 92.5276 |
| test03.fasta.gz | 8.71E-01 | GCF_021432085.1_genomic.fna.gz | 4775870 | 90.7837 |
| test03.fasta.gz | 8.76E-01 | GCF_003935375.1_genomic.fna.gz | 4657537 | 90.5424 |
| test03.fasta.gz | 8.78E-01 | GCF_000341615.1_genomic.fna.gz | 4674664 | 90.4745 |
| test03.fasta.gz | 8.79E-01 | GCF_014764705.1_genomic.fna.gz | 4861582 | 90.4108 |
| test03.fasta.gz | 8.79E-01 | GCF_000935215.1_genomic.fna.gz | 4878963 | 90.398 |
| test03.fasta.gz | 8.82E-01 | GCF_002929225.1_genomic.fna.gz | 4898053 | 90.2678 |
| test03.fasta.gz | 8.83E-01 | GCA_007713455.1_genomic.fna.gz | 4711826 | 90.2361 |
| test03.fasta.gz | 8.86E-01 | GCF_003696315.1_genomic.fna.gz | 4321164 | 90.1098 |
Query_Name是您的查询基因组,Distance是基因组Jaccard距离(1-J/Jp),Neighbor_Fasta_name是基于基因组Jaccard距离的最近邻,按距离排序。ANI是按照上述公式从基因组Jaccard距离计算得出的,用于您查询基因组与最近数据库基因组的距离。
我们还提供了一些脚本,用于分析请求的输出并将其与其他基于ANI的方法进行比较:[https://github.com/jianshu93/gsearch_analysis](https://github.com/jianshu93/gsearch_analysis)
Ann
对于UMAP-like算法执行降维并在基因组数据库中可视化,我们在tohnsw步骤(预建数据库)后运行它(见下面ann使用部分)。有关详细信息,请参阅[annembed](https://github.com/jean-pierreBoth/annembed)。然后可以可视化这一步骤的输出,例如,对于GTDB v207,我们有以下图表。请参阅[论文](https://www.biorxiv.org/content/10.1101/2024.01.28.577627v1)。
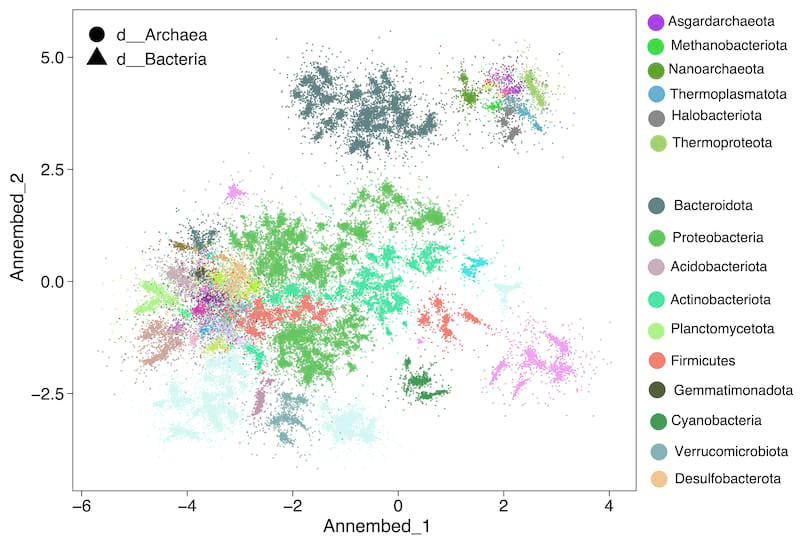
数据库拆分
我们提供了一组脚本,允许将数据库基因组拆分为N部分,并为每一部分构建HNSW图数据库,然后针对这些部分运行新基因组的搜索并收集结果。这只需要您机器的1/N内存,但代价是相同的查询基因组需要额外的sketching。
### for a folder with genomes in it, we can split it into N subfolders randomly by running:
./scripts/split_folder.sh input_folder_path output_folder_path 3
### using the output from above split_folder.sh step, build database for each subfolder
./scripts/multiple_build.sh output_folder_path gsearch_db_folder
### using output from above multiple_build.sh step, search new genomes againt each database
./scripts/multiple_search.sh gsearch_db_folder_output new_genome_folder_path output
简单安装案例
预建二进制文件将在发布页面[https://github.com/jean-pierreBoth/gsearch/releases/tag/v0.1.3-beta](https://github.com/jean-pierreBoth/gsearch/releases/tag/v0.1.3-beta)上提供,适用于主要平台(无需安装,只需下载并使其可执行)。我们建议您使用此发布页面上的Linux版本(GSearch_Linux_x86-64_v0.1.3.zip),以便方便(仅需要系统库)。对于macOS,我们建议使用对应平台的mac-binaries(GSearch_Darwin_x86-64_v0.1.3.zip或GSearch_Darwin_aarch64_v0.1.3.zip)。或者,对于Windows,请使用GSearch_pc-windows-msvc_x86-64_v0.1.3.zip。
或者,如果您在Linux上安装了conda
![]()
### we suggest python 3.8, so if you do not have one, you can create a new one
conda create -n python38 python=3.8
conda activate python38
conda config --add channels bioconda
conda install gsearch -c bioconda
#### a different binary name was used for bioconda channel, you can change it to gsearch later, see below
gsearchbin -h
### change to gsearch binary name
cp $(which gsearchbin) $CONDA_PREFIX/bin/gsearch
gsearch -h
或者您在macOS上安装了homebrew
brew tap jianshu93/gsearch
brew update
brew install gsearch
gsearch -h
否则,您可以通过自行安装/编译来完成(参见安装部分)
### get the binary for linux (make sure you have recent Linux installed with GCC, e.g., Ubuntu 18.0.4 or above)
wget https://github.com/jean-pierreBoth/gsearch/releases/download/v0.1.3-beta/GSearch_Linux_x86-64_v0.1.3.zip --no-check-certificate
unzip GSearch_Linux_x86-64_v0.1.3.zip
## get the x86-64 binary for macOS
wget https://github.com/jean-pierreBoth/gsearch/releases/download/v0.1.3-beta/GSearch_Darwin_x86-64_v0.1.3.zip --no-check-certificate
unzip GSearch_Darwin_x86-64_v0.1.3.zip
## get the aarch64/arm64 binary for macOS
wget https://github.com/jean-pierreBoth/gsearch/releases/download/v0.1.3-beta/GSearch_Darwin_aarch64_v0.1.3.zip --no-check-certificate
unzip GSearch_Darwin_aarch64_v0.1.3.zip
## get the binary for Windows, ann subcommand is not available for windows for now
wget https://github.com/jean-pierreBoth/gsearch/releases/download/v0.1.3-beta/GSearch_pc-windows-msvc_x86-64_v0.1.3.zip
## Note that for MacOS, you need sudo previlege to allow external binary being executed
* **make it excutable (changed it accordingly on macOS)**
chmod a+x ./gsearch
### put it under your system/usr bin directory (/usr/local/bin/ as an example) where it can be called
mv ./gsearch /usr/local/bin/
### check install
gsearch -h
### check install MacOS, you may need to change the system setup to allow external binary to run by type the following first and use your admin password
sudo spctl --master-disable
使用方法
gsearch -h
************** initializing logger *****************
Approximate nearest neighbour search for microbial genomes based on MinHash-like metric
Usage: gsearch [OPTIONS] [COMMAND]
Commands:
tohnsw Build HNSW graph database from a collection of database genomes based on MinHash-like metric
add Add new genome files to a pre-built HNSW graph database
request Request nearest neighbors of query genomes against a pre-built HNSW graph database/index
ann Approximate Nearest Neighbor Embedding using UMAP-like algorithm
help Print this message or the help of the given subcommand(s)
Options:
--pio <pio> Parallel IO processing
--nbthreads <nbthreads> nb thread for sketching
-h, --help Print help
-V, --version Print version
##我们在此提供使用预建数据库的利用示例。
### download neighbours for each genomes (fna, fasta, faa et.al. are supported) as using pre-built database, probminhash or SetSketch/hll database (prob and hll)
wget http://enve-omics.ce.gatech.edu/data/public_gsearch/GTDBv207_v2023.tar.gz
tar -xzvf ./GTDBv207_v2023.tar.gz
### Densified MinHash database (optdens) for NCBI/RefSeq, go to https://doi.org/10.6084/m9.figshare.24617760.v1
### Download the file in the link by clicking the red download to you machine (file GSearch_GTDB_optdens.tar.gz will be downloaded)
wget http://enve-omics.ce.gatech.edu/data/public_gsearch/GSearch_optdens.tar.gz
mkdir GTDB_optdens
mv GSearch_GTDB_optdens.tar.gz GTDB_optdens/
### get test data, we provide 2 genomes at nt, AA and universal gene level
wget https://github.com/jean-pierreBoth/gsearch/releases/download/v0.0.12/test_data.tar.gz --no-check-certificate
tar xzvf ./test_data.tar.gz
###clone gsearch repo for the scripts
git clone https://github.com/jean-pierreBoth/gsearch.git
### test nt genome database
cd ./GTDB/nucl
##default probminhash database
tar -xzvf k16_s12000_n128_ef1600.prob.tar.gz
# request neighbors for nt genomes (here -n is how many neighbors you want to return for each of your query genome, output will be gsearch.neighbors.txt in the current folder)
gsearch --pio 100 --nbthreads 24 request -b ./k16_s12000_n128_ef1600_canonical -r ../../test_data/query_dir_nt -n 50
## reformat output to have ANI
../../gsearch/scripts/reformat.sh 16 1 ./gsearch.neighbors.txt ./clean_ANI.txt
## SetSketch/hll database
tar -xzvf k16_s4096_n128_ef1600.hll.tar.gz
gsearch --pio 100 --nbthreads 24 request -b ./k16_s4096_n128_ef1600_canonical_hll -r ../../test_data/query_dir_nt -n 50
## reformat output to have ANI
../../gsearch/scripts/reformat.sh 16 1 ./gsearch.neighbors.txt ./clean_ANI.txt
## Densified MinHash, download first, see above
cd GTDB_optdens
tar -xzvf GSearch_GTDB_optdens.tar.gz
cd GTDB
#nt
tar -xzvf k16_s18000_n128_ef1600_optdens.tar.gz
gsearch --pio 100 --nbthreads 24 request -b ./k16_s18000_n128_ef1600_optdens -r ../../test_data/query_dir_nt -n 50
## reformat output to have ANI
../../gsearch/scripts/reformat.sh 16 1 ./gsearch.neighbors.txt ./clean_ANI.txt
### or request neighbors for aa genomes (predicted by Prodigal or FragGeneScanRs), probminhash and SetSketch/hll
cd ./GTDB/prot
tar xzvf k7_s12000_n128_ef1600.prob.tar.gz
gsearch --pio 100 --nbthreads 24 request -b ./k7_s12000_n128_ef1600_gsearch -r ../../test_data/query_dir_aa -n 50
## reformat output to have AAI
../../gsearch/scripts/reformat.sh 16 1 ./gsearch.neighbors.txt ./clean_AAI.txt
#aa densified MinHash
cd ./GTDB_optdens
cd GTDB
tar -xzvf k7_s12000_n128_ef1600_optdens.tar.gz
gsearch --pio 100 --nbthreads 24 request -b ./k7_s12000_n128_ef1600_optdens -r ../../test_data/query_dir_aa -n 50
## reformat output to have AAI
../../gsearch/scripts/reformat.sh 16 1 ./gsearch.neighbors.txt ./clean_ANI.txt
### or request neighbors for aa universal gene (extracted by hmmer according to hmm files from gtdb, we also provide one in release page)
cd ./GTDB/universal
tar xzvf k5_n128_s1800_ef1600_universal_prob.tar.gz
gsearch --pio 100 --nbthreads 24 request -b ./k5_n128_s1800_ef1600_universal_prob -r ../../test_data/query_dir_universal_aa -n 50
###We also provide pre-built database for all RefSeq_NCBI prokaryotic genomes, see below. You can download them if you want to test it. Following similar procedure as above to search those much larger database.
##graph database based on probminhash and setsketc/hll, both nt and aa
wget http://enve-omics.ce.gatech.edu/data/public_gsearch/NCBI_RefSeq_v2023.tar.gz
##nt graph database based on densified MinHash, go to below link to download GSearch_k16_s18000_n128_ef1600_optdens.tar.gz
https://doi.org/10.6084/m9.figshare.24615792.v1
#aa graph database, densified MinHash, go to below link to download GSearch_k7_s12000_n128_ef1600_optdens.tar.gz
https://doi.org/10.6084/m9.figshare.22681939.v1
### Building database. database is huge in size, users are welcome to download gtdb database here: (<https://data.ace.uq.edu.au/public/gtdb/data/releases/release207/207.0/genomic_files_reps/gtdb_genomes_reps_r207.tar.gz>) and here (<https://data.ace.uq.edu.au/public/gtdb/data/releases/release207/207.0/genomic_files_reps/gtdb_proteins_aa_reps_r207.tar.gz>) or go to NCBI/RefSeq to download all available prokaryotic genomes
### build database given genome file directory, fna.gz was expected. L for nt and .faa or .faa.gz for --aa. Limit for k is 32 (15 not work due to compression), for s is 65535 (u16) and for n is 255 (u8)
gsearch --pio 2000 --nbthreads 24 tohnsw -d db_dir_nt -s 12000 -k 16 --ef 1600 -n 128 --algo prob
gsearch --pio 2000 --nbthreads 24 tohnsw -d db_dir_aa -s 12000 -k 7 --ef 1600 -n 128 --aa --algo prob
### When there are new genomes after comparing with the current database (GTDB v207, e.g. ANI < 95% with any genome after searcing, corresponding to >0.875 ProbMinHash distance), those genomes can be added to the database
### old .graph,.data and all .json files will be updated to the new one. Then the new one can be used for requesting as an updated database
gsearch --pio 100 --nbthreads 24 add -b ./k16_s12000_n128_ef1600_canonical -n db_dir_nt (new genomes directory)
### or add at the amino acid level, in the parameters.json file you can check whether it is DNA or AA data via the "data_t" field
cd ./GTDB/prot
gsearch --pio 100 --nbthreads 24 add -b ./k7_s12000_n128_ef1600_gsearch -n db_dir_nt (new genomes directory in AA format predicted by prodigal/FragGeneScanRs)
### visuzlizing from the tohnsw step at amino acid level (AAI distance), output order of genome files are the same with with seqdict.json
cd ./GTDB/prot
gsearch ann -b ./k7_s12000_n128_ef1600_gsearch --stats --embed
输出解释
gsearch.neighbors.txt是您当前目录中的默认输出文件。
对于查询_dir中的每个基因组,都会找到请求的N个最近基因组,并按距离(从小到大)或ANI(从大到小)排序。如果查询中的某个基因组在输出文件中不存在,意味着在此级别(nt或aa),数据库中没有这样的最近邻(或者距离数据库中最佳匹配很远),您可以然后转到氨基酸水平或通用基因水平。
依赖项、特性和安装
特性
-
hnsw_rs依赖于crate simdeez来加速距离计算。在Intel上,您可以使用simdeez_f特性构建hnsw_rs。
-
annembed依赖于openblas,因此您必须在“annembed_openblas-static”、“annembed_openblas-system”或“annembed_intel-mkl”这三个特性之间进行选择。您可能需要安装gcc、gfortran和make。这可以通过下面的选项进行,或者通过修改Cargo.toml中的特性部分来实现。在这种情况下,只需填写您想要的默认值。
-
kmerutils提供了一个名为“withzmq”的特性。该特性可以用于在服务器上存储压缩质量并运行请求。在这个crate中不是必需的。
自行安装/编译
首先安装Rust工具
curl --proto '=https' --tlsv1.2 -sSf https://sh.rustup.rs | sh
从Crates.io安装和编译gsearch(推荐)
- 简单安装,启用annembed时将使用intel-mkl
cargo install gsearch --features="annembed_intel-mkl"
或使用系统安装的openblas
cargo install gsearch --features="annembed_openblas-system"
- 在MacOS上,需要动态库链接(您必须首先安装openblas):(注意,openblas安装的lib路径在M1 MACs上不同)
所以您需要运行
brew install openblas
echo 'export LDFLAGS="-L/usr/local/opt/openblas/lib"' >> ~/.bash_profile
echo 'export CPPFLAGS="-I/usr/local/opt/openblas/include"' >> ~/.bash_profile
echo 'export PKG_CONFIG_PATH="/usr/local/opt/openblas/lib/pkgconfig"' >> ~/.bash_profile
source ~/.bash_profile
cargo install gsearch --features="annembed_openblas-system"
从github的最新版本安装gsearch
- 从github直接安装
cargo install gsearch --features="annembed_intel-mkl" --git https://github.com/jean-pierreBoth/gsearch
- 下载和编译
git clone https://github.com/jean-pierreBoth/gsearch
cd gsearch
#### build
cargo build --release --features="annembed_openblas-static"
cargo build --release --features="annembed_intel-mkl"
###on MacOS, which requires dynamic library link (install openblas first, see above):
cargo build --release --features="annembed_openblas-system"
文档生成
可以通过运行以下命令生成HTML文档(以下为使用“annembed_openblas-system”特性的人的示例)
cargo doc --features="annembed_openblas-system" --no-deps --open
然后安装FragGeneScanRs
cargo install --git https://gitlab.com/Jianshu_Zhao/fraggenescanrs
如果出现问题(包括在ARM64 CPU上安装/编译),以下是一些提示这里
预构建数据库
我们为细菌/古菌、病毒和真菌提供了预构建的基因组/蛋白质组数据库图形文件。蛋白质组数据库是基于每个基因组中的基因,由FragGeneScanRs(https://gitlab.com/Jianshu_Zhao/fraggenescanrs)为细菌/古菌/病毒和GeneMark-ES版本2(http://exon.gatech.edu/GeneMark/license_download.cgi)为真菌预测的。
- 细菌/古菌基因组是GTDB数据库的最新版本(v207,67,503个基因组)(https://gtdb.ecogenomic.org),在95% ANI定义了细菌物种。请注意,GSearch还可以运行更高分辨率的物种数据库,如99% ANI。
- 截至2023年1月,NCBI/RefSeq的细菌/古菌基因组(~318,000个基因组)(https://www.ncbi.nlm.nih.gov/refseq/about/prokaryotes/),没有在给定的ANI阈值上进行聚类。
- 病毒数据库基于JGI IMG/VR数据库的最新版本(https://genome.jgi.doe.gov/portal/IMG_VR/IMG_VR.home.html),该数据库也在95% ANI定义了病毒OTU (vOTU)。
- 真菌数据库基于整个RefSeq真菌基因组(通过MycoCosm网站检索),我们进行了去重,并在99.5% ANI定义了真菌物种。
- 所有四个预构建数据库都可以在这里找到:http://enve-omics.ce.gatech.edu/data/gsearch或FigShare,请参阅下载链接这里
待办事项列表
1.C-MinHash,具有循环排列的单排列哈希(Li和Li 2022 ICML),在准确性方面是最佳的MinHash,比任何其他MinHash-like哈希在RMSE方面都有显著改进。使用循环排列为从OPH生成的sketch向量中的空桶,以便空桶可以借用到来自其他非空桶的哈希值,而不仅仅是复制最近的非空桶,就像在密集MinHash方案中那样。然而,对于大D和常规K(MinHash K)的循环排列向量可能很大,它必须进行K次循环移动并应用K次,这可能会消耗大量的内存。对于微生物基因组,D通常为10^7左右,K为10^4到10^5以获得99% ANI的准确性,排列向量不是问题。
2. UltraLogLog(Ertl 2023) 和 ExaLogLog (Ertl 2024),在基数估计的空间/内存方面对 HyperLogLog 有显著改进。在获得两个集合的基数后,尽管存在较大的方差,我们也可以使用包含和排除规则来估计 Jaccard 指数。一个简单但高效的基数估计新估计器,FGRA(Further Generalized Remaining Area),基于 Tau-GRA(Wang 和 Pettie,2023),可以被使用。但我们也可以将 UltraLogLog 映射到相应的 HyperLogLog,使用最大似然估计(MLE)进行基数(较慢),或联合最大似然估计(JMLE)进行交集基数(然后是 Jaccard 指数),它们比 FGRA 稍微更准确,并满足 Cramér-Rao 下界。ExaLogLog 估计器也是基于 MLE,但由于空间要求较小,它并不那么快。然而,类似于 Hyperloglog 的方案不在局部敏感哈希方案下,RMSE 比类似于 MinHash 的方案大得多。
3. 对于极其庞大的基因组数据库,例如 10^9 或更多的基因组,sketch 的大小和 HNSW 图的大小线性增加,这对存储/加载如此大的文件构成了挑战。然而,仅对大型基因组数据库的小部分进行 sketch 和构建 HNSW 图是可能的/容易的,并创建几个小的 sketch 文件和 HNSW 图文件用于搜索。我们可以逐个搜索这些几个小部分,然后收集搜索结果(最近邻)并根据 Jaccard 距离对它们进行排序。这在算法上等同于(对于准确性和运行时间)为整个大型数据库进行 sketch 和构建 HNSW 图,但只需占用一小部分内存。但如何选择这个小部分以便计算效率高?如果以流式的方式实现,Coreset 算法可以用来将相似的基因组聚类到簇中,这与 k-medoid 类似但计算效率显著更高,请参阅 coreset crate 此处。我们肯定会尽快实现这一想法。现在,将大型数据库手动分割成小部分很容易,类似于这个想法在图基 NNS 库中的实际工业级应用 此处
参考文献
-
赵建书,Jean Pierre-Both,Luis M. Rodriguez-R 和 Konstantinos T. Konstantinidis,2022. GSearch:通过结合 Kmer Hashing 与分层可导航小世界图进行快速且可扩展的微生物基因组搜索。 bioRxiv 2022:2022.2010.2021.513218. biorxiv.
-
赵建书,Jean Pierre-Both,Konstantinos T Konstantinidis,2024. 近似最近邻图提供快速有效的嵌入,并在大规模生物数据中的应用。 bioRxiv 2024.01.28.577627. biorxiv
依赖关系
~22–61MB
~1M SLoC