31个稳定版本 (5个主要版本)
| 8.2.0 | 2024年3月21日 |
|---|---|
| 7.2.0 | 2024年3月3日 |
| 6.3.3 | 2024年2月2日 |
| 6.2.0 | 2023年9月12日 |
| 3.0.1 | 2023年6月25日 |
#919 in 科学
在 3 crates 中使用
150KB
3.5K SLoC
BLUTILS
Blutils 工具允许用户运行并生成Blast结果的共识身份。目前BlastN可用。
安装
Blutils 包可以从 crates.io 使用 cargo 直接安装
cargo install blutils-cli
安装后,可以使用 blu 命令调用 Blutils。
blu --help
输出应接近以下内容
A utility to make it easier to run and analyze Blast results
Usage: blu <COMMAND>
Commands:
build-db Build the blast database as a pre-requisite for the blastn command
blastn Execute the parallel blast and run consensus algorithm
check Check `Blutils` dependencies
help Print this message or the help of the given subcommand(s)
Options:
-h, --help Print help information
-V, --version Print version information
检查依赖项
在运行 Blutils 之前,您可以可选地检查操作系统依赖项。自然地,BLutils 依赖于在宿主系统上安装的 Ncbi-Blast+ 工具以执行并行 blast 搜索。要检查宿主操作系统是否已安装这些软件包,请在 Linux 系统上运行 Blutils 检查器
blu check linux
注意:目前系统检查仅适用于 Linux 系统,并假定可以从终端直接调用依赖项。
使用 Blutils 运行 Blast
Blutils 的执行很简单。要检查所有可用选项,调用 blast 子命令的帮助
blu blastn run-with-consensus --help
检查可用选项后,使用测试数据运行 Blutils。首先从项目的 GitHub 目录下载测试数据
export INPUT_DIR=https://raw.githubusercontent.com/sgelias/blutils/main/test/mock/input
curl ${INPUT_DIR}/query/query.fna > query.fna
curl ${INPUT_DIR}/query/ref_databases/mock-16S.fna > mock-16S.fna
curl ${INPUT_DIR}/query/ref_databases/mock-16S_taxonomies.tsv > mock-16S_taxonomies.tsv
然后运行 Blutils
blu blastn run-with-consensus \
query.fna \
mock-16S.fna \
mock-16S_taxonomies.tsv \
output \
-t 6 \
--taxon bacteria \
--strategy relaxed \
-f
似乎上述命令,输出文件可以在输出目录中找到,其中将包含两个附加文件,分别命名为 blast.out 和 blutils.consensus.json。第一个包含默认的 Blast 表格响应,而后者是 Blutils 的响应,将接近以下内容:
[
{
"query": "NR025123.135626.Bac",
"taxon": {
"rank": "species",
"identifier": "shewanella-olleyana",
"percIdentity": 100.0,
"bitScore": 2695.0,
"alignLength": 1459,
"mismatches": 0,
"gapOpenings": 0,
"qStart": 1,
"qEnd": 1459,
"sStart": 1,
"sEnd": 1459,
"eValue": 0.0,
"taxonomy": "d__bacteria;p__pseudomonadota;c__gammaproteobacteria;o__alteromonadales;f__shewanellaceae;g__shewanella;s__shewanella-olleyana",
"mutated": true,
"consensusBeans": null
}
},
{
"query": "draft-5123",
"taxon": {
"rank": "species",
"identifier": "bacillus-mojavensis-subgroup",
"percIdentity": 100.0,
"bitScore": 815.0,
"alignLength": 441,
"mismatches": 0,
"gapOpenings": 0,
"qStart": 1,
"qEnd": 441,
"sStart": 217,
"sEnd": 657,
"eValue": 0.0,
"taxonomy": "d__bacteria;clade__terrabacteria-group;p__bacillota;c__bacilli;o__bacillales;f__taxid-186817;g__bacillus;species-group__bacillus-subtilis-group;species-subgroup__bacillus-mojavensis-subgroup",
"mutated": true,
"consensusBeans": [
{
"rank": "species",
"identifier": "bacillus-halotolerans",
"occurrences": 5
},
{
"rank": "species",
"identifier": "bacillus-mojavensis",
"occurrences": 4
}
]
}
},
{
"query": "INVALID_SEQUENCE",
"taxon": null
}
]
Blast 执行
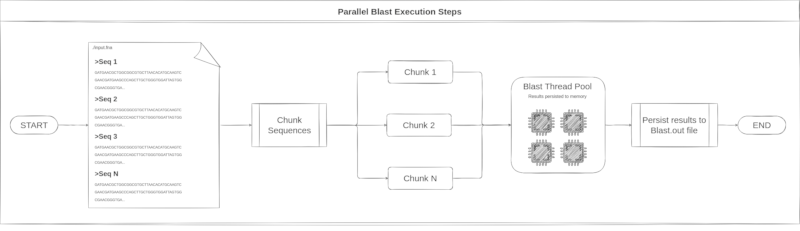
Blast 执行试图达到可用的全部 CPU 利用率。在默认的多线程 blast 执行模式下,无法达到完全饱和。通过 Blutils 运行 Blast 是可能的。在此过程中所采取的所有步骤都可以在下面的图像中看到。
共识生成
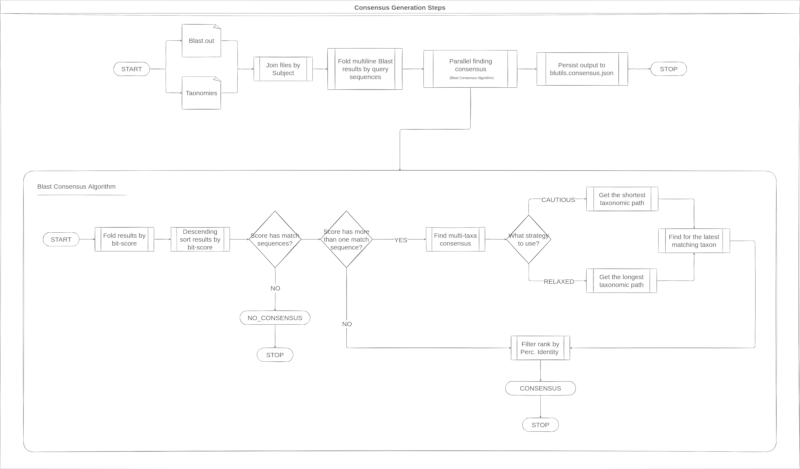
与来自QIIME 2的共识生成不同,Blutils共识算法根据Blast结果对位得分和百分比身份进行数据预过滤,看起来就像下面图片中描述的算法。
下一步
本项目仅包含运行BlastN和生成共识身份的基本功能。因此,需要创建许多其他功能,如创建数据库提取器从官方NCBI taxdump结果获取数据并同时在构建FASTA数据库,以及其他功能。我们欢迎在需要时提出新功能建议!
依赖项
~23–34MB
~590K SLoC